Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Histone deacetylases (HDACs)
3. Therapeutic strategies...
4. Perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(3):424-442. doi:10.7150/ijms.30154 This issue Cite
Review
Histone Deacetylases (HDACs) Guided Novel Therapies for T-cell lymphomas
Qing Zhang1 ![]() , Shaobin Wang2, Junhui Chen1, Zhendong Yu3
, Shaobin Wang2, Junhui Chen1, Zhendong Yu3 ![]()
1. Department of Minimally Invasive Intervention, Peking University Shenzhen Hospital, Shenzhen, Guangdong, 518036, China.
2. Health Management Center of Peking University Shenzhen Hospital, Shenzhen, Guangdong, 518036, China.
3. China Central Laboratory of Peking University Shenzhen Hospital, Shenzhen, Guangdong, 518036, China.
Received 2018-9-24; Accepted 2018-12-19; Published 2019-1-29
Abstract
T-cell lymphomas are a heterogeneous group of cancers with different pathogenesis and poor prognosis. Histone deacetylases (HDACs) are epigenetic modifiers that modulate many key biological processes. In recent years, HDACs have been fully investigated for their roles and potential as drug targets in T-cell lymphomas. In this review, we have deciphered the modes of action of HDACs, HDAC inhibitors as single agents, and HDACs guided combination therapies in T-cell lymphomas. The overview of HDACs on the stage of T-cell lymphomas, and HDACs guided therapies both as single agents and combination regimens endow great opportunities for the cure of T-cell lymphomas.
Keywords: Histone deacetylases (HDACs), T-cell lymphomas, cutaneous T-cell lymphoma, peripheral T-cell lymphoma, combination therapy
1. Introduction
1.1 T-cell lymphomas
T-cell lymphomas encompass a heterogeneous group of malignancies comprising 15-20% of systemic lymphomas and less than 10% non-Hodgkin lymphomas. T-cell malignancies are endemic in East Asia, the Caribbean, intertropical Africa, the Middle East, South America, and Papua New Guinea. Adult T-cell lymphoma caused by human T-cell leukemia virus-1 is one of the most popular T-cell malignancies. Up to now, there are about 5-20 million patients infected with HTLV-1 around world. According to WHO/EORTC classification based on the clinical pathology indications, T-cell malignancies are divided into extra nodal T cell lymphoma, cutaneous T cell lymphomas (Sézary syndrome and Mycosis fungoides), anaplastic large cell lymphoma, and angioimmunoblastic T cell lymphoma [1]. The clinical performance and pathogenesis are divergent between different T-cell lymphomas; the therapeutic strategies are different but with some overlap especially in their early stage. However, because of the comprehensive pathogenesis and limited therapeutic strategies, patients with T-cell lymphomas usually have a poor prognosis and easy to relapse.
1.2 Current therapeutic strategies for T-cell lymphomas
Currently, chemotherapy is the most preferred treatment in patients with T-cell lymphoma. With the development of intensified chemotherapy, the prognosis of T-cell leukemia/lymphoma has gradually improved. However, due to the non-specific toxicity of current chemotherapeutic compounds to normal cells, the progressive loss of quality of life is a matter of concern [2]. Besides, patients with relapse and resistance to conventional chemotherapy exhibit limited efficacy in the salvage setting.
Recently, the booming targeted therapeutic strategies have become the forefront therapeutics in patients with severe lymphomas and achieved promising therapeutic effects, such as immune therapies of antibodies against CTLA 4 and PD 1 /PDL 1 [3], B cell receptor signaling pathway inhibitors (e.g., ibrutinib [4]), NK cells (e.g., AFM13 [5]), bispecific T engager (BiTE, blinatumomab [6]), T cell receptor (e.g., CART19 [7, 8]). However, all these gorgeous strategies mentioned above have not been testified or not suitable in the treatment of T-cell lymphomas.
The treatment of patients with T-cell lymphomas is challenging. Novel therapeutic strategies are urgently warranted to improve the therapeutic effects of T-cell lymphomas. Thus new flourishing therapeutic targets and techniques (drugs, techniques or combinational trials) would provide opportunities for the treatment of patients with T-cell malignancies. HDACs, kinases, T/B cell receptors, checkpoints or other related key modulators are promising choices. In this review, we will focus on the role of HDACs in T-cell lymphomas and the potential applications as therapeutic targets for the treatment of patients with T-cell malignancies.
2. Histone deacetylases (HDACs)
2.1 Brief profile of HDACs
Histone deacetylases are a class of enzymes deacetylating the acetyl group from ε-N-acetyl lysine amino acid from a histone, which lead to the tight wrapping of DNA. HDACs play a key role in the homeostasis maintenances of histone acetylation euchromatin and heterochromatin in living system [9, 10]. There are 18 HDACs recognized up to now, divided into HDAC I (HDAC 1, 2, and 3: mainly in the nucleus; HDAC 8: partially in the cytoplasm), HDAC II (HDAC 4, 5, 6, 7, 9, and 10: shuttling in and out of the nucleus), as Table 1 showed [11]. Considering the main function of the process of de-acetylation, HDACs act as key epigenetic modulators of essential biological processes by modifying histones of chromatin or non-histone proteins (PTEN, APE1/Ref-1 (APEX1), NF-κB, aggressomes, et al.) [12]. Besides, HDACs are involved in protein degradation, especially HDAC6 that was reported to interfere with HSP90 via degrading HSP90-interacting proteins [13, 14]. HDACs are thought to be the main pathogenic factors to both leukemia and other solid tumors, such as chronic myeloid leukemia, chronic lymphocytic leukemia[15], pediatric acute myeloid leukemia, acute promyelocytic leukemia, renal cancer, colorectal and gastric cancer, breast tumors and so on[16]. In this review, we have focused on the role of HDACs in T-cell lymphomas and the application of HDAC inhibitors as T-cell lymphomas treatments.
Classification of histone deacetylases (HDACs)
| Cofactor | Class | HDAC members | Localization |
|---|---|---|---|
| Zn2+--dependent | Class I | HDAC 1,2,3,8 | Nucleus (HDAC 8, partially in the cytoplasm) |
| Class IIA | HDAC 4,5,7,9 | Nucleus/cytoplasm | |
| Class IIB | HDAC 6,10 | Mainly in the cytoplasm | |
| Class IV | HDAC 11 | Nucleus/cytoplasm | |
| NAD+--dependent | Class III | SIRT 1,6,7 | Nucleus |
| SIRT 2 | Cytoplasm | ||
| SIRT 3,4,5 | Mitochondria |
Current statuses of HDAC inhibitors applied in T-cell lymphomas.
| Name | Chemical structure | Activity | Disease | Adverse effects | Status | Administration | Ref | |
|---|---|---|---|---|---|---|---|---|
| Enzyme | IC50/nM | |||||||
| Vorinostat | 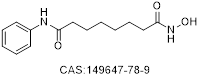 | Class I Class II | < 86 | R/R CTCL | Common side effects | US FDA Japan Europe | Oral | [37-44] |
| Romidepsin | 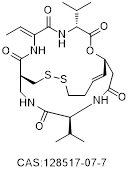 | HDAC 1 | 36 | CTCL R/R PTCL | Acceptable | US FDA | Intravenous | [59-74] |
| HDAC 2 | 47 | |||||||
| Belinostat | 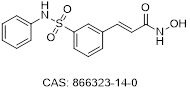 | Class I Class II Class IV | Nano molar potency | R/R PTCL | Acceptable | US FDA | Intravenous | [17, 135, 136] |
| Chidamide | 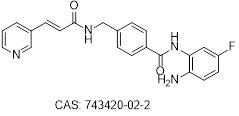 | Class I : 1,2,3 | R/R PTCL | Grade 1 to 2 | CFDA | Oral | [31, 32, 51, 91-96] | |
| Class II b: 10 | ATL PTCL | Clinical trial US Japan | ||||||
| Panobinostat | 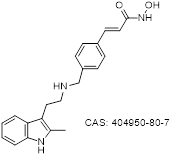 | Class I Class II Class IV | Pan-HDACi | ATL CTCL PTCL | Common side effects | Phase II | Oral | [105, 107-121, 123, 124, 137] |
| Remetinostat | 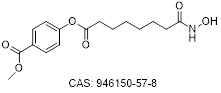 | CTCL | No systemic AE | Phase II | Medivir AB | |||
| Entinostat | 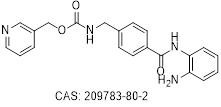 | HDAC1 | 510 | ATL | Toxic | Preclinical | [39] | |
| HDAC3 | 1700 | |||||||
2.2 Modes of action in T-cell lymphoma
The elaboration of HDACs in the pathogenesis, therapy and prognosis of T-cell lymphomas would help a lot in the treatment of patients with T-cell lymphomas. The precise mechanisms of HDACs in T-cell malignancies have been investigated under the intervention of HDAC inhibitors (Figure 1), but still not been fully elucidated.
Molecular functions of HDACs. This figure was adapted from Bodiford & Reddy (2014)[17], Hood & Shah (2016)[18], and Weiguo Zhu (2014)[19].
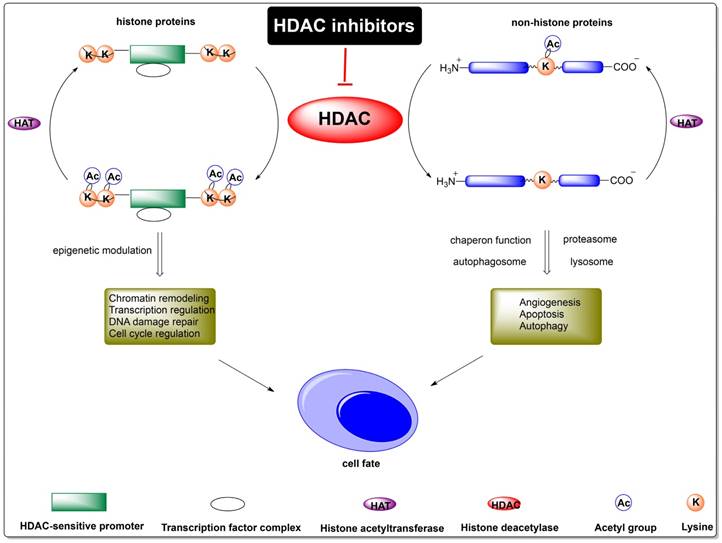
2.2.1 Epigenetic modulation
Except for genetic mutation, chromatin differentiation, the reversible epigenetic alterations are the main alterations for cancer initiation, progression and invasion [20]. HDACs interfere with epigenetic chromatin modification by de-acetylating histones and non-histone proteins of nuclear transcription factors mediating carcinogenesis [21]. Besides, HDACs participate in the mediation of oncogenes of Bcl-xL-[22], Bcl2-[23], or TCRβ [22], c-Myc [24], Notch3 [25].
2.2.2 Cytokines regulation
Cytokines are the important participators in immune regulation. A response rate of 30% for cutaneous T-cell lymphoma (CTCL) expressing high affinity IL-2 receptor (IL-2R) was demonstrated in Phases I and II clinical trials of DAB (389) IL-2 by inhibition of histone deacetylases (HDACs) interfering with 0.06 mM arginine butyrate[26]. Up-regulation of HDAC1 and HDAC6 and transcriptional induction of the conco-miR-21 were found in CTCL, which was caused by excessive autocrine secretion of IL-15 in T-cells. IL-15 mediated inflammation was critical in CTCL, and a novel oncogenic regulatory loop between IL-15, HDAC 1/6, and miR-21 was discovered [27]. Besides, HDAC deactivation and concomitant down regulation of CD27 were found in TEL-AML positive leukemia by CD40 ligation [28]. Under the improvement of immune surveillance by CD40 ligation in patients with TEL-AML, a potential increased relapse-free survival was observed with the presence of other risks.
2.2.3 Apoptosis
HDACs mediated the pro-apoptotic response in a decreased way and increased the apoptotic threshold in various hematological and solid lymphomas. The inhibition of HDAC 1/2 was found to have a synergism effect with BCL11B (a key T-cell development regulator) in the anti-apoptosis effect in CTCL lines [29]. HDACs promoted the acetylation of the chaperone heat shock protein 90 (HSP90), leading to the binding and stabilization of HSP90 client proteins RASGRP1 and CRAF which activate the mitogen-activated protein kinase pathway signaling and down-regulate the pro-apoptotic BCL2 family member BIM both in vitro and in vivo. siRNA of RASGRP1, HSP90 inhibition or HDAC inhibition may contribute to the cytotoxicity and apoptosis induction in lymphoid malignancies by influencing the signal pathway related to HSP90, RASGRP1, CRAF, and BIM [30]. HDAC inhibition by Chidamide induced cell apoptosis by down-regulating Bcl-2 and up-regulating cleaved Caspase-3 and Bax protein expression in peripheral T cell lymphoma [31], MDS cell lines (SKM-1, MUTZ-1) and AML cell line (KG-1) [32]. The induction of tumor suppressor gene RhoB, pronounced expression of p21 and global CpG methylation in the modulation of HDAC were found in CTCL cell lines and tumor cells derived from Sézary syndrome patients by the combined treatment of HDAC inhibitor Romidepsin and demethylating agent Azacitidine [33]. HDAC also has a negative correlation with an anti-apoptotic drug resistance related molecule surviving [34], which was demonstrated by HDAC inhibitor SAHA in the treatment of ATL cells. Considering the important regulation of HDAC in the downstream signal pathway and apoptotic related proteins, HDACs may serve as potential drug target for T-cell malignancies in an apoptosis induction way.
2.2.4 Autophagy
Autophagy is a key self-salvage process to various stresses by degrading long-lived proteins and damaged organelles. Except for the main function on lysine de-acetylation in chromatin, HDACs also have functions in the regulation of plethora of cytosolic proteins with different cellular functions (such as angiogenesis, immune responses, and autophagy) in series of cancers. Treatment of T-cell lymphomas and other cellular studies with effective HDAC inhibitors induced apoptosis, cell-cycle arrest, cell differentiation, anti-angiogenesis and autophagy. In CTCL, HDAC inhibitor SAHA up-regulated the expression of autophagic factor LC3, and inhibited the mammalian target of rapamycin (mTOR) leading to activation of autophagic protein kinase ULK1 [35]. Except for T-cell lymphomas, HDACs were related to autophagy in other cancers. The novel, potent, selenium-containing HDAC inhibitors (SelSA-1 and SelSA-2) were demonstrated to simultaneously increase in autophagy in lung cancer A549 cells [36]. HDACs have a synergistic effect with autophagy in the process of cellular survival, thus autophagy targeting and HDACs inhibiting offer alternative choice for the treatment of T-cell lymphomas.
HDAC inhibitors usually act at the transcriptional level by interfering with epigenetic chromatin modification and histone de-acetylation [21]. HDAC inhibitors have been shown to induce the acetylation of histone and non-histone proteins (nuclear transcription factors mediating anti-cancer activity), cause DNA damage, promote the re-expression of repressed genes during oncogenesis, mediate lethality through cytokinesis failure, facilitate a pro-apoptotic response and lower the cellular apoptotic threshold in various hematological and solid lymphomas. By modulating these key physiological and pathological processes related to apoptosis, immune response, autophagy and metabolism, HDACs play indispensable role in the pathogenesis and prognosis of T-cell malignancies, which make them prospective target for the treatment of T-cell malignancies.
3. Therapeutic strategies targeting HDACs
3.1 Single agent strategies
The specific functions and structures of HDACs make them ideal drug targets. Various HDAC inhibitors that differ in potency, selectivity, gene regulation, and non-histone protein targets have been investigated. Most HDAC inhibitors have similar mechanism of actions against HDAC by binding to the zinc atom in the catalytic pocket in a non-competitive manner. Nowadays, plenty of HDAC inhibitors have been developed as promising anti-cancer agents; several HDAC inhibitors are now in clinical trials; some have been approved as single anti-tumor agents or adjuvants to traditional chemotherapeutics for many cancers. As for T-cell lymphomas, especially peripheral T-cell lymphoma (PTCL) and cutaneous T-cell lymphoma (CTCL), four classes of HDAC inhibitors have been developed. FDA has approved several HDAC inhibitors for the treatment of T-cell lymphomas: Vorinostat (SAHA) for CTCL, Romidepsin for CTCL and PTCL, and Belinostat for PTCL; and Chidamide has been approved by the CFDA for the treatment of relapsed/refractory TCL in China as a single agent. The development of HDAC inhibitors for the treatment of T-cell lymphomas is now flourishing, many of which exhibit excellent potential in HTLV-1-infected cell lines, ATL cell lines, freshly isolated primary ATL cells, PTCL, and CTCL, such as Romidepsin (cyclic depsipeptide FR901228), Panobinostat (LBH-589), trichostatin A (TSA), and benzamide MS-275. More and more optimization of these reported HDAC inhibitors are now under investigation. In this part, we summarized reported HDAC inhibitors, which have been approved for the treatment of T-cell lymphomas or demonstrated potential efficacy in T-cell lymphomas, as Figure 2 showed.
Chemical structures of HDAC inhibitors.

3.1.1 Vorinostat (suberoylanilide hydroxamic acid: SAHA)
Vorinostat (Zolinza®), also called SAHA, is a hydroxamic acid discovered from screening a series of bishydroxamic acids [37, 38]. Further studies to optimize of the molecular structure and determine the mechanism of action of SAHA showed that by directly binding to the catalytic pocket of HDACs, SAHA inhibited the enzymatic activities of both class I and II HDACs, with an IC50 of less than 86 nM [37, 38]. The cyclic structure of SAHA may be the key point for its specificity. In cellular studies, SAHA effectively inhibited the proliferation of human mantle cell lymphoma cells, human T-cell lymphotropic virus type I (HTLV-1)-infected T cells (MT-1, MT-2, MT-4, and HUT102), established CTCL cell lines, freshly isolated ATL cells and circulating malignant CTCL cells harvested from patients by up-regulating of P21 waf1 protein [39], decreasing the level and phosphorylation of STAT6 protein, and increasing the ratio of NF-κB in the cytoplasm versus the nucleus, leading to growth arrest and apoptosis [40]. Based on its specificity towards HDACs and excellent performance in T-cell lymphomas, SAHA was tested in a multicenter clinical trial in patients with refractory and relapsed CTCL, and showed excellent therapeutic potential, with an ORR of 24% [41] (30% [42]) and duration of response of approximately 4 months [41] (longer than 6 months [42]) in two single-arm phase II studies. In an extension study, 32% of overall patients experienced improving pruritus relief and high quality of life. Based on these promising therapeutic effects, Vorinostat was first approved as an HDAC inhibitor in 2006 for the treatment of relapsed/refractory cutaneous T-cell lymphoma (CTCL) patients with recurrent disease on or after 2 systemic therapies as an oral agent named Vorinostat in USA [43] and Japan and achieved orphan drug designation in Europe. Vorinostat is now the only approved drug for the treatment of relapsed/refractory CTCL with a daily recommended dose of 400 mg [44]. According to reports, glucuronidation by the uridine diphosphate glucuronosyl-transferase (UGT) enzyme system was identified as the key process mediating the metabolism and excretion of Vorinostat, while the cytochrome P-450 isoenzyme system was not found to participate in the metabolism of Vorinostat [45]. UGT1A1 might play an important role in Vorinostat toxicity and response levels in related patients. Until now, warfarin and valproic acid have demonstrated obvious drug interactions with Vorinostat, which should be noticed in the clinic [46]. Vorinostat shows a favorable safety profile and well tolerance at the approved once-daily dose of 400 mg. The most common adverse events of Vorinostat are of grade 1 or 2 (such as fatigue, nausea, and diarrhea), while more severe toxicities (such as thrombocytopenia, fatigue, and nausea) only occur in less than 6% of patients [47, 48]. Except for R/R CTCL, Vorinostat has exhibited promising therapeutic effects and manageable safety profiles [49] against other hematologic lymphomas (such as multiple myeloma (MM) [50], indolent NHL[51, 52], relapsed/refractory acute myeloid leukemia (AML) [53, 54], myelodysplastic syndrome (MDS)) [50, 54, 55], glioblastoma multiforme [56, 57], advanced leukemias [50], and solid tumors [58] as a monotherapy or as a combination therapy with other agents (e.g., PIs and IMiDs) at doses tolerated by patients. The optimization of the structure of Vorinostat and studies on its efficacy in other lymphomas are now under investigation.
3.1.2 Romidepsin (cyclic depsipeptide FR901228)
Romidepsin (also called FR901228) is a natural product isolated from the bacterium Chromobacterium violaceum; this product has a typical cyclic depsipeptide structure, and primarily displays an inhibitory effect on class I HDACs and a weak effect on class IIB (HDAC 6) [59, 60]. This inhibition was found to have potent effects on T-cell lymphomas. Romidepsin acts as a prodrug, and its mode of action was elucidated in 1998 [61]. The key interaction of Romidepsin with HDAC is it's binding to the zinc atom in the binding pocket of Zn-dependent histone deacetylase after reducing the disulfide in cells [62]. Romidepsin has shown excellent anti-cancer effects by interfering with the cell cycle, cell motility and angiogenesis, thus inducing cell death and differentiation. Romidepsin exhibited effective durable responses in patients with relapsed/refractory CTCL as a single-agent therapy. The FDA approved Romidepsin in 2009 for the treatment of cutaneous T-cell lymphoma (CTCL) patients who have received at least 1 prior systemic therapy [63, 64]. Besides, it has shown excellent inhibitory effects on malignant lymphoid cell lines, including HTLV-1-infected T-cell lines, primary adult T-cell leukemia and peripheral T-cell lymphoma (PTCL) cells by blocking the Notch 1 pathway [65] and the NF-ĸB pathway [66]. Romidepsin has demonstrated durable clinical responses in patients with relapsed/refractory PTCL [64, 67-72], leading to its approval by the FDA in June 2011 for the treatment of PTCL in patients who have received at least one prior therapy. Combination therapies of Romidepsin with other agents are now in clinical trials, for example, Romidepsin plus CHOP is in a phase Ib/II trial of patients with newly diagnosed PTCL (NCT01280526, NCT01796002) [73], and a combination therapy using Romidepsin and pralatrexate has shown synergy in preclinical studies[74]. All these ongoing trials have shown the potential of Romidepsin in the treatment of PTCL. In conclusion, Romidepsin exhibits outstanding effects on the treatment of CTCL and PTCL as a single agent and has potential as a single agent or in combination with other effective agents for the treatment of T-cell lymphomas. It would be worthwhile to further explore the application and optimization of the chemical structure of Romidepsin to improve its efficiency and safety/tolerability.
3.1.3 Belinostat (PXD101)
Belinostat (PXD101), N-hydroxy-3-[3-(phenysulfamoyl) phenyl] prop-2-enamide, is a low-molecular-weight pan-HDAC inhibitor with a sulfonamide-hydroxamide structure developed by TopoTarget [75]. Belinostat exhibits nanomolar potency towards class I, II and IV HDAC isoforms by chelating hydroxamate with zinc ions essential for the enzymatic activity of HDAC. Belinostat has demonstrated meaningful efficacy and a favorable toxicity profile towards serious hematological lymphomas and solid tumors [76, 77]. The encouraging efficacy of Belinostat in T-cell lymphoma has accelerated its development as a therapeutic agent [17, 18]. In clinical trial of patients with relapsed/refractory PTCL [78, 79], Belinostat exhibited promising efficacy and a highly favorable safety profile (minimal grade 3 and grade 4 toxicity) [80]. In addition, this inhibitor is especially well tolerated in patients with thrombocytopenia and shows promising benefits. Based on these conclusive therapeutic effects, Belinostat (BELEODAQ™, Spectrum Pharmaceuticals, Inc.) has been accelerated approved by the FDA in July 2014 as an orphan drug and fast track designed for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL) by intravenous administration[81, 82]. The safety and efficacy of Belinostat has made it a first-line drug for R/R PTCL, and combination treatments using Belinostat with other front-line therapies are now in clinical trials, which will be further elucidated in the section on combination therapies in this review. Other than the use of Belinostat in the treatment of PTCL, the clinical application of Belinostat in solid tumor lymphomas [76, 79, 83-85], refractory acute leukemia [84, 86, 87], myelodysplastic syndrome [88], and nonsmall-cell lung cancer [89, 90] has also been further evaluated. In any case, these results guarantee the expansion of applying Belinostat in cancer therapy and offer more therapeutic choices for PTCL. This novel HDAC inhibitor may thus represent a breakthrough in the treatment of T-cell lymphomas and other HDAC-related solid cancers.
3.1.4 Chidamide (HBI-8000)
Chidamide (HBI-8000 or CS055) is a synthetic analog of MS-275 screened from various benzamide-prototype compounds and further rationally designed by molecular docking employing an HDAC-like protein, which was independently developed by Chipscreen Biosciences in China as an innovative new drug. In the following chemical genomic-based analyses and other molecular biological evaluations, Chidamide exhibited subtype-selectivity towards class I HDACs (HDAC 1, 2, 3) and class IIb HDAC (HDAC 10) by targeting the catalytic pocket [91]. Cellular assays showed the efficient anti-cancer activity of Chidamide in ATL-derived cell lines, primary ATL cells, myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) cell lines in a time- and dose-dependent manner by increasing histone H3 acetylation at Lys9/Lys18 and H4 at Lys8 [91], immune cell (NK cells and CD8 cells)-mediated cytotoxicity [91], epigenetic modulation [31], cell cycle arrest at G0/G1 phase [31, 32, 92], apoptosis by JAK2/STAT3 [93] and Bim and NLR family pyrin domain containing 3 (NLRP3) inflammasome pathway activation [94]. Based on its efficiency in ATL cells, Chidamide was further developed as an anti-tumor agent in PTL patients, especially in treatment-naive or relapsed patients. In addition, its efficacy and safety have been further demonstrated in non-Hodgkin's lymphoma in phase I and II clinical trials, with an overall response rate (ORR) of 39.06, disease control rate (DCR) of 64.45% and median progression-free survival (PFS) of 129 (95% CI 82 to 194) days as a single agent [51, 95, 96]. After oral administration in patients, Chidamide showed excellent potential in overcoming drug resistance, tumor cell metastasis, and recurrence by inducing tumor stem cell differentiation, drug-resistant tumor cell reversal, and epithelial-to-mesenchymal transition [31]. The adverse events (AEs) of Chidamide were mostly grade 1/2, while more than 5% patients showed grade 3 or even higher adverse events (such as thrombocytopenia in 10.2% of patients and neutropenia in 6.2% of patients) after receiving Chidamide monotherapy [96]. Encouraging preliminary anti-tumor activity, favorable PK and PD profiles, and safety profiles of Chidamide were demonstrated in serial clinical trials, especially in patients with T-cell lymphomas [31]. The China Food and Drug Administration (CFDA) as an oral agent for the treatment of relapsed or refractory PTCL approved Chidamide in December 2014. Long-term survival potential was observed in PTCL patients after treated with Chidamide. Moreover, clinical trials are currently ongoing in the United States (Clinical Trials Identifier: NCT02733380) and Japan for ATL/PTCL patients. In addition to ATL/PTCL, Chidamide also exhibited a broad-spectrum of therapeutic effects against other hematological lymphomas [92] and solid cancers (such as lung [97, 98], colon [99, 100], liver [101] and pancreatic [102-104] carcinoma) as shown in athymic nude mice subcutaneously inoculated with different human tumor cell lines. Further investigation of its potential efficacy, safety profile and mechanism as a single agent or combination therapy in T-cell lymphomas and solid tumors is now underway.
3.1.5 Panobinostat (LBH-589)
Panobinostat (LBH589) is a novel cinnamic hydroxamic acid derivative with excellent inhibitory activity against class I (HDAC 1, 2, 3, and 8), II (HDAC 4, 5, 6, 7, 9, 10), and IV (HDAC 11) HDAC enzymes, and is defined as a pan-deacetylase inhibitor [105]. As a pan-DAC inhibitor, Panobinostat exhibited at least 10-fold more potency than other HDAC inhibitors (such as Vorinostat) and appears to be the most potent pan-DAC inhibitor. In vitro, Panobinostat has shown potent cytotoxicity at nanomolar LD90 (90% cell death, 14-541 nM) in many hematological lymphomas, such as multiple myeloma [106-109], WM cells and cell lines [110], acute myelogenous leukemia [111], and cutaneous T-cell lymphoma [112-115]. Panobinostat was approved in February 2015 for the treatment of relapsed/refractory MM patients who had gone through at least two prior regimens, including Bortezomib and an immunomodulatory agent in combination with Bortezomib and Dexamethasone by the US FDA and the European Medical Association (EMA). Studies of the anti-tumor effects of Panobinostat in T-cell lymphoma, such as cutaneous T-cell lymphoma, adult T-cell leukemia/lymphoma cells, are ongoing. The anti-tumor potential of Panobinostat has been attributed to the inhibition of angiogenesis and migration [116], disruption of endothelial cell chemotaxis, and induction of apoptosis [117] and autophagy [118]. Panobinostat was shown to induce hyperacetylation of core histone proteins of the chromatin such as histone H3 and H4, deregulation of Polycomb repressive complex 2 (PRC2) by over-expressing Enhancer of zeste homolog 2 (EZH2) [119, 120], SUZ12 [119], EED and CCR6, downregulation of miR-150 and miR-185-5p in advanced CTCL [112], modulation of epigenetic genes, reactivation of epigenetically silenced tumor suppressor genes (such as p21 and TP53 [121]), and regutation of signaling pathways (Akt [122], Sirt1 [114], NF-κB, Bcl-2 and STAT3/STAT5 [114]). The clinical efficacy of Panobinostat by oral and IV administration has been demonstrated in advanced cutaneous T-cell lymphoma (CTCL) [113]. In a phase I dose-escalation study of oral Panobinostat, two patients achieved CR and four achieved PR among ten CTCL responding patients, while dose-limiting diarrhea and thrombocytopenia were observed [115]. Panobinostat exhibited acceptable tolerability and modest overall clinical responses in CTCL patients with a manageable safety profile. Microarray analyses showed that a unique set of genes related to apoptosis, immune regulation, and angiogenesis were altered after Panobinostat treatment [115]. A phase II trial of oral Panobinostat in patients with refractory cutaneous T-cell lymphoma (CTCL) failed at least two systemic therapies with relatively low response rates and short time to progression (CRs: 15 of 95 patients) [123]. In a phase II trial of oral Panobinostat in a total number of 139 patients with MF/SS refractory to 2 standard therapies, the median TTRs was 2.3 months in bexarotene-exposed group (n=79), while 2.8 months in bexarotene-naïve group (n=60); the median DORs of bexarotent -exposed group was 5.6 months; the median PFS rates of bexarotent -exposed group was 4.2 months and 3.7 months for bexarotene-naïve group [113]. Patients with PTCL-NOS and CD4+ hematodermic T-cell lymphoma exhibited potential responses after being treated with Panobinostat (NCT00901147) [124]. The most common adverse effects of Panobinostat were diarrhea, nausea, fatigue, pruritus, thrombocytopenia, and decreased appetite. The potential study of Panobinostat in advanced or refractory CTCL have been promoted into phase II clinical trials as a single agent or combination treatment. Besides, Panobinostat is currently being evaluated in an ongoing clinical study for peripheral T-cell lymphoma [125]. Although Panobinostat exhibited excellent anti-tumor potential in T-cell lymphoma patients with acceptable tolerance and a manageable safety profile as an oral agent, based on the completed clinical trials, any government institute has not yet approved it. Further investigations and more clinical trials of Panobinostat in T-cell lymphomas as a single agent or in combination with other anti-tumor agents are in full swing. In any case, the properties of Panobinostat in T-cell lymphomas (anti-tumor potency, safety profile, tolerance, oral formulation, long half-life, etc.) have made it an attractive alternative therapeutic agent for T-cell lymphomas, especially cutaneous T-cell lymphoma and adult T-cell leukemia/lymphoma.
3.1.6 Remetinostat
Remetinostat is a class of benzoic acid targeting HDACs, which was developed by Medivir AB for the treatment of early-stage cutaneous T-cell lymphoma (CTCL). Unlike other systemic HDAC inhibitors, remetinostat was designed to be active within cutaneous lesions and to be quickly decomposited in the bloodstream, preventing exposure to the whole body. Based on this specificity, remetinostat exhibits high efficacy in the skin with mild side effects. Medivir AB has recently announced that remetinostat, the topical skin-directed histone deacetylase (HDAC), has completed a 60-subject phase II clinical study in patients with an early-stage mycosis fungoides (MF) variant of CTCL in which remetinostat showed good tolerance without signs of systemic adverse effects in all dose groups. The full phase II trial data will be presented at scientific meeting in the second half of 2017. Based on the efficacy and safety data from this phase II study, Medivir expects to carry out a phase III study later this year after discussing the data and protocol with regulatory authorities. The promising therapeutic benefits and safety make remetinostat a promising therapeutic treatment of patients with CTCL, a chronic and poorly treated orphan disease.
3.1.7 Entinostat (MS-275)
Entinostat (SNDX-275 or MS-275) is a synthetic benzamide derivative showing activity against HDAC 1 (IC50=0.51 μM) and HDAC 3 (IC50=1.7 μM) and anti-tumor activity in solid cancers (bladder cancer, metastatic kidney cancer, non-small cell lung cancer, and myeloid lymphomas) and lymphomas (e.g., B-cell chronic lymphocytic leukemia [126]). Entinostat has been tested in many clinical trials in patients with advanced and refractory solid tumors or lymphoma [127-130] as a single agent or in combination with other agents, such as in metastatic kidney cancer (Clinical Trials Identifier: NCT01038778), relapsed and refractory myeloid lymphomas (phase II study, Clinical Trials Identifier: NCT00466115), non-small cell lung cancer (Clinical Trials Identifier: NCT00387465) [131-133]. Entinostat has been demonstrated to effectively inhibit the proliferation of both human T-cell lymphotropic virus type I (HTLV-1)-infected T cells (MT-1, MT -2, MT -4, and HUT102) and freshly isolated ATL cells harvested from patients by interfering NF-κB signaling and inducing apoptosis [39]. Although entinostat has exhibited exciting anti-tumor potency, its toxic effect cannot be ignored. Thus, in order to benefit from its potency as an anti-tumor agent, more efforts should be made in order to improve its toxicity via optimization of its chemical structure.
In addition to the HDAC inhibitors mentioned above that have been studied intensively, many other exciting HDAC inhibitors have been developed, such as PCI-24781 (abexinostat), ITF- 2357 (givinostat), MGCD 0103 (mocetinostat), FK228 (Romidepsin), and valproic acid [134]. These HDAC inhibitors have exhibited promising potency towards HDAC enzymes and therapeutic effects as single agents or adjuvants to existing therapeutic strategies.
3.2 Combinational therapies
On the way to the discovery of ant-cancer therapies, combination therapies have occupied considerable markets in clinical. Combination therapies take advantages of many effective therapies (such as chemotherapies, targeting therapies, and immune therapies) and synergistic effects were demonstrated in patients with malignancies. Upon regulating related histone and non-histone proteins, HDACs participated in serious cellular processes, proliferation, epigenetic modulation, cytokine secretion, apoptosis, autophagy, signal transduction network, immune modulation, and so on. We have introduced the effective HDAC inhibitors as single agents for the treatment of T-cell lymphomas aforementioned. Considering the participation of HDACs in complex cellular processes, the combination therapies of HDACs inhibition and others against T-cell lymphomas are worth to explore and verify elaborately. The combination of HDAC inhibitors and other anticancer agents may exhibit synergistic efficacy in the treatment of T-cell lymphomas. However, reviews on HDAC-guided combination therapies for T-cell lymphomas are sparse. In this section, we have gone through the potential HDAC-guided combination therapies in the treatment of T-cell lymphomas both preclinical and clinical. Data regarding combination treatments with HDACIs is sparse.
3.2.1 Belinostat based combinational therapies
Belinostat (PXD101) is a pan-HDAC inhibitor with a sulfonamide-hydroxamide structure developed by TopoTarget[138]. Belinostat has become a first-line drug for R/R PTCL based on its high efficacy and acceptable safety profile. CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) or CHOP-like strategies are recommended as the first-line treatment for PTCL, but the prognosis remains poor with a high possibility of relapsing within 5 years. Belinostat and components of the CHOP strategy have different cellular targets and mechanisms of action, there will be a great chance that Belinostat and CHOP- or CHOP- like strategy has a synergistic effect against patients with PTCL. A phase I study of 23 patients with PTCL was carried out for the investigation of effect of Belinostat and CHOP (NCT01839097)[139]. In this study, a response rate of 89% (16 of 18 evaluable patients) was demonstrated upon the treatment of Belinostat (standard therapeutic doses) and CHOP, and the adverse events were those typically reported with CHOP alone, such as neutrophil count decreased (26%), anemia (22%), neutropenia (17%) and white blood cell count decreased (17%). A study of Belinostat and Bortezomib in treating patients with relapsed or refractory acute leukemia or myelodysplastic syndrome has been completed (NCT01075425). A study of Belinostat plus Carfilzomib in relapsed/refractory NHL (non-Hodgkin lymphoma, diffuse large B-cell lymphoma, mantle cell lymphoma, follicular lymphoma, peripheral T-cell lymphoma) is currently recruiting participants (NCT02142530). A study of volasertiv and Belinostat in patients with relapsed and refractory aggressive B-cell and T-cell lymphomas is waiting for the participant recruitment (NCT02875002). Another study of Belinostat therapy with Zidovudine for adult T-cell leukemia-lymphoma is currently recruiting participants (NCT02737046).
All these clinical trials of Belinostat (Table 3) with other agents ongoing or completed for the treatment of patients with T/B cell lymphomas or other hematological malignancies or other solid tumors exhibited the potential therapeutic effect of Belinostat in these malignanices, providing alternative options for patients with malignancies and opportunities for doctors and drug researchers.
Belinostat based combination therapies.
| Agent1 | Agent2 | T-cell lymphomas | Progress | Clinical trial | Ref. |
|---|---|---|---|---|---|
| Belinostat | CHOP | R/R PTCL | Phase I | NCT01839097 | [132] |
| Carfilzomib | R/R NHL (including PTCL) | Phase I | NCT02142530 | ||
| Bortezomib | Acute leukemia or myelodysplastic syndrome | Phase I | NCT01075425 | ||
| Volasertiv | R/R T/B-cell lymphomas | Phase I | NCT02875002 | ||
| Zidovudine | Adult T-cell leukemia-lymphoma | Phase I | NCT02737046 |
Vorinostat based combination therapies.
| Agent1 | Agent2 | T-cell lymphomas | Progress | Clinical trial | Ref. |
|---|---|---|---|---|---|
| Vorinostat | 5-Aza-2'deoxycytidine | CTCL | Preclinical | ||
| Gemcitabine/Busulfan/Melphalan | Refractory/poor-risk relapsed lymphomas | Phase I | NCI201102891 | [133] | |
| Rituximab | R/R indolent NHL | Phase II | [135] | ||
| CHOP | Newly diagnosed PTCL | Phase I | [136] | ||
| IFN α and extracorporeal photopheresis | Mycosis fungoides and Sézary syndrome | Preclinical | [137] | ||
| PI3K inhibitors or HSP90 inhibitors | Cutaneous T-cell lymphoma cells | Preclinical | [138, 139] | ||
| ABT-737 | Human T-lymphotropic virus type-I (HTLV-1) infected T-cell lines and fresh ATL cells | Preclinical | [17] | ||
| Bexarotene | CTCL | Preclinical | [140] | ||
| Bortezomib | CTCL | Preclinical | [141] | ||
| Lenalidomide/Dexamethasone | R/R PTCL | Phase I | [142] | ||
| UVASens/UV-A photochemotherapy | CTCL Myla cell line | Preclinical | [143] |
Romidepsin based combination therapies.
| Agent1 | Agent2 | T-cell lymphomas | Progress | Clinical trial | Ref. |
|---|---|---|---|---|---|
| Romidepsin | CHOP | PTCL | Phase III | NCT01280526; NCT01796002 | [144] |
| ICE (Ifosfamide, Carboplatin and Etoposide) | Phase I | NCT01590732 | [145] | ||
| Lenalidomide | R/R lymphomas (including TCL) | Phase I/II | NCT01755975; NCT02341014; NCT02232516; NCT01755975 | [146, 147] | |
| Alisertib | TCL cell lines, R/R aggressive B/T-cell lymphomas | Preclinical, phase I | NCT01897012 | [148] | |
| Azatidine | CTCL cell lines and fresh Sézary syndrome cells | Preclinical | [27] | ||
| Gemcitabine | R/R PTCL | Phase II | [150] | ||
| Pralatrexate | TCL cell lines and a NOG murine model of TCL | Preclinical | [68] | ||
| γ-secretase inhibitor compound E and bortezomib | A murine MT-1 model of human ATL | Preclinical | [59] | ||
| LY2409881 | Lymphoma cells | Preclinical | [60] | ||
| Low dose localized electron beam radiation therapy (LEBT) | CTCL | Phase I | [152] | ||
| Interferon gamma | Preclinical | [153] | |||
| Poly ICLC, radiation | Advanced CTCL | Phase I | NCT02061449 |
Chidamide, Panobinostat, AN-7, and MS-275 based combination therapies.
| Agent1 | Agent2 | T-cell lymphomas | Progress | Clinical trial | Ref. |
|---|---|---|---|---|---|
| Chidamide | Chemotherapy | R/R PTCL | Phase I | [92] | |
| Doxorubicin | PTCL cells | Preclinical | [154] | ||
| Panobinostat | Everolimus | R/R lymphoma (including TCL) | Phase I | NCT00967044 | [157] |
| Bortezomib | PTCL | Phase III | NCT01023308 | [129, 158] | |
| AN-7 | Doxorubicin | MF/SS cell lines, SPBL, T-cell lymphoma | Preclinical | [160] | |
| MS-275 | UVASens/UV-A photochemotherapy | CTCL Myla cell line | Preclinical | [143] |
3.2.2 Vorinostat (SAHA) based combination therapies
Preclinical experiments of Vorinostat with the inhibition of methyltransferase or proteasome, or with DNA-damaging agents (radiation or chemicals induced) have demonstrated promising synergistic activity in specific tumor types. After treating with Vorinostat, the cellular DNA methyltransferase was up regulated, which could be preclinically abrogated by DNA methyltransferase inhibition. This synergistic effect could increase the lethality of tumor cells, offering new opportunity for combination therapy in tumors. A clinical study of 60 patients with refractory or poor-risk relapsed lymphoma (26 with diffuse large B-cell lymphoma (DLBCL), 21 with Hodgkin lymphoma, 8 with T-cell lymphoma, and 5 with other B-cell lymphomas) was conducted to test the clinical combination effect of Azacitidine with Vorinostat/Gem/Bu/Mel (NCI201102891) [140]. In the follow-up of 15 months, patients with T-cell lymphoma have 88% of event-free and overall survival rates observed after treating with the combination therapy. The event-free and overall survival rates are very promising in other patients with lymphomas (65% and 77% among patients with DLBCL, 76% and 95% among patients with Hodgkin lymphoma). In another clinical study of 78 patients (52 DLCL, 20 HL, and 6 T-lymphoma) between ages 12 to 65, 5 of 6 patients with T-NHL were alive in CR at 16 to 29 months after treating with the combination therapy at the median follow-up 25 months [141]. The main toxicities included mucositis, dermatitis, neutrophils and platelets, with no treatment-related deaths. This clinical trial of Vorinostat and Gemcitabine and Busulfan and Melphalan demonstrated high efficacy and acceptable safety profile in refractory/poor-risk relapsed lymphomas. But further evaluation is still needed when applied widely in clinical. Vorinostat was demonstrated to down regulate nuclear factor kappa beta (NFκβ), leading to the increasing of Rituximab activity. A phase II study was conducting in 28 patients with newly diagnosed or relapsed/refractory indolent NHL to investigate the effect of the combination of Vorinostat and Rituximab [142]. After orally administration of Vorinostat (200 mg twice a day on days 1-14) and Rituximab (375mg/m2 on day 1 per 21- day cycle), the ORR of all patients was 46% (67% in the newly diagnosed and 41% in relapsed/refractory patients), and the median PFS was 29.2 months (18.8 months for relapsed/refractory and not reached for untreated patients). A phase I study of Vorinostat with standard CHOP in patients with newly diagnosed peripheral T-cell lymphoma was also undertaken and good therapeutic effects were gotten [143].
The study of Vorinostat in combination with IFN α and extracorporeal photopheresis in the progressive and/or stable disease of progressive and treatment refractory late stage mycosis fungoides and Sézary syndrome exhibited prolonged efficacy of responses [144]. Vorinostat modifies signaling of T-cell receptor, MAPK, JAK-STAT pathways, and dephosphorylate kinases of ZAP70 (Tyr319, Tyr493) and its downstream target AKT (Ser473). Combination of Vorinostat with PI3K inhibitors or HSP90 inhibitors resulted synergic cytotoxic antagonism in cutaneous T-cell lymphoma cells, which would show potential efficacy for the treatment of T-cell lymphomas [145, 146]. Combination of Vorinostat with the Bcl-2 homology 3 mimetic ABT-737 which blocks Bcl-2, Bcl-XL, and Bcl-w in human T-lymphotropic virus type-I (HTLV-1) infected T-cell lines and fresh ATL cells induced increasing apoptosis and decreasing expression of survivin, offering new choice for the treatment of ATL [23]. The retinoid X receptor (RXR)-agonist bexarotene is a monotherapeutic regimen for cutaneous T-cell lymphomas (CTCLs), and the combination of bexarotene and HDAC inhibitor Vorinostat in vitro leads to synergetic activation of gene transcription and increasing of cell death in human tumor cell lines, but only with low doses of each drug relative to the product label [147]. The proteasome inhibitor Bortezomib also has a synergistic interaction with Vorinostat in cutaneous T cell lymphoma by synergistically inducing apoptosis, providing a rationale for clinical trials of combined proteasome and histone deacetylase inhibition in the treatment of CTCL [148]. Not all the combination therapy of Vorinostat has potential therapeutic effect in T cell lymphomas. In a study of combination of Lenalidomide, Vorinostat and Dexamethasone in 8 patients with relapsed/refractory peripheral T cell lymphoma (PTCL) (2 peripheral T cell lymphoma unspecified, 5 angioimmunoblastic T cell lymphoma, 1 ALK-negative anaplastic large-cell lymphoma) [149]. The median progression-free survival and OS was only 2.2 and 6.7 months separately. The poor therapeutic effects have not terminated the application in T cell lymphomas, but need further investigations with other combination regimens.
HDACs play an important role in the stabilization of DNA double strand. UVASens/UV-A phototherapy induced DNA strand breakage by abstraction of H-atoms from 1-deoxyribosyl carbons. In a preclinical study of CTCL Myla cell line, HDAC inhibitors Vorinostat and MS-275 significantly augment UVASens/UV-A photochemotherapy-induced cytotoxicity [150]. Further animal models and clinical studies are needed to further evaluate the combinatorial effects of this combination.
3.2.3 Romidepsin related combination therapies
Multiple clinical trials were undertaken in T-cell lymphomas to evaluate the synergestic therapeutic effect of combination therapies with Romidepsin.
Chemotherapy is the first-in line therapy for patients with T-cell lymphomas. The combination of Romidepsin with conventional anti-leukemic agents (Methotrexate, Vincristine, Imatinib, Cytarabine, Carboplatin, Doxorubicin, 4-hydroperoxy-cyclophosphamide, Etoposide, 6-mercaptopurine and SN-38) has exihibited additive effects in T/B-cell lymphomas, making Romidepsin a promising candidate for the treatment of T/B-cell lymphomas as combination therapies [151]. A phase Ib/II study of Romidepsin and CHOP in patients with newly diagnosed PTCL was evaluated (NCT01280526) [73, 151]. In this study, the combination exhibited excellent therapeutic effect with an ORR of 69%, estimated PFS of 41% and OS of 71% at the median follow-up of 30 months. The most common adverse effects were grade ≥ 3 hematologic and all-grade non-hematologic (such as gastrointestinal, respiratory, or general conditions). A separate phase III study of the comparison of CHOP vs Romidepsin plus CHOP in patients with untreated PTCL is currently recruiting participants (NCT01796002). In the investigation of Romidepsin with ICE (Ifosfamide, Carboplatin and Etoposide), the therapeutic effective was positive with an ORR of 78% (CR: 64%), the median PFS of 10.0 months, OS of 12.5 months, and common grade 3/4 adverse effects of thrombocytopenia and neutropenia (NCT01590732) [152].
Romidepsin has also shown synergistic effects with Lenalidomide in TCL cell lines and relapsed/refractory lymphomas [153], and a few clinical studies have been conducted. In a phase I/II study of relapsed/refractory lymphomas and multiple myeloma (MM), an ORR of 67% in patients with TCL and adverse effects of pneumonia and thrombocytopenia exhibited. In another analysis of 21 TCL patients, the ORR reached 53% (CR: 11%), and the most common AEs were grade ≥ 3 (neutropenia, thrombocytopenia, anemia, and electrolyte abnormalities)[154]. There have been many other clinical trials of Romidepsin and Lenalidomide in the treatment of patients with T-cell lymphomas are undergoing, such as NCT01755975, NCT02341014, NCT02232516, NCT01755975.
Aurora kinase inhibitor alisertib exhibited promising therapeutic effect against TCL as a single-agent. The combination of Romidepsin and alisertib has shown highly synergistic effect in TCL cell lines through modulation of cytokinesis [155]. A phase I clinical trial of this combination is currently ongoing in relapsed/refractory aggressive B/T-cell lymphomas (NCT01897012) [156]. The therapeutic effect was well in patients with PTCL (1 CR and 1 SD in patients with PTCL up to now), and this therapeutic combination may tend to be more effective in patients with PTCL.
There is a potential that the combination of Romidepsin and other epigenetic therapies would benefit for the treatment of patients with T-cell lymphomas. The combination of Romidepsin and Azatidine exerted synergistic anti-proliferative effects by inducing apoptosis and changing the global CpG methylation profile in CTCL cell lines and fresh tumor cells isolated from Sézary syndrome patients [33].
In a phase II trial of investigation of the combination of Gemcitabine plus Romidepsin in patients with relapsed/refractory PTCL, the ORR was 30% (CR: 15%), the two-year survival was 50% and progression-free survival was 11.2% [157]. All these patients under investigation were enduring grade ≥3 adverse events (including thrombocytopenia (60%), neutropenia (50%), and anemia (20%)) and mild and transient non-hematological toxicities. The synergy was observed in this trial, but not as prominent as in preclinical phase. The combination of Romidepsin and pralatrexate exhibited a concentration-dependent synergism against a panel of TCL cell lines in vitro and enhanced efficacy in a NOG murine model of TCL comparing to any single agent, endowing the future clinical application in patients with TCLs [74].
Romidepsin was found to activate the MAPK pathway in HuT78 cutaneous T-cell lymphoma (CTCL) cells, one of the reasons of HDAC inhibitor resistance [158]. Romidepsin also participate in the regulation of chaperone proteins Hsp90, Hsp70 and transcription factors C-MYC and p53. All these comprehensive regulation mechanism make Romidepsin a potential combination regimen. In certain ATL patients, the activity of HDACs, Notch-1 signaling and nuclear factor-κB pathway were increased and constitutive activated. The combination of HDAC inhibitor Romidepsin, γ-secretase inhibitor compound E, and the proteasome inhibitor Bortezomib exhibited significantly enhanced antitumor efficacy in a murine MT-1 model of human ATL, providing rational supports for further clinical trial of this combination therapy strategy in the treatment of patients with ATL [65]. A novel IKK2 inhibitor LY2409881 efficiently suppressed the activity of the NF-κB subunit p65 in lymphoma cells pretreated with Romidepsin, underlying the potential of the combination of Romidepsin and IKK2 inhibitor in the treatment of patients with B/T-cell lymphomas [66]. Romidepsin has also showed synergistic effect with low dose localized electron beam radiation therapy (LEBT) in patients with CTCL, laying foundation for the formal clinical trial of this combination in patients with CTCL [159]. The combination of Romidepsin and interferon gamma may exhibit stabilization of Th1 cytokine profiles and enhanced cytotoxic T-cell activity resulting to a more robust anti-tumor response [160]. A clinical study of the combination of poly ICLC, radiation, and Romidepsin in the treatment of advanced cutaneous T cell lymphoma is currently recruiting participants (NCT02061449).
In addition to the specific treatment of T-cell lymphomas, the combinations of Romidepsin and other therapies have been investigated in other lymphoid malignancies (cutaneous or peripheral T-cell lymphomas included). Many clinical trials for the evaluation of Romidepsin related combination therapy are now recruiting participants, such as Romidepsin plus oral 5-Azacitidine in relapsed/refractory lymphoid malignancies (NCT01998035), Romidepsin and pralatrexate in relapsed/refractory lymphoid malignancies (NCT01947140), Romidepsin, Gemcitabine, Oxaliplatin, and Dexamethasone in relapsed/refractory aggressive lymphomas (NCT02181218).
3.2.4 Chidamide based combination therapies
Chidamide (CS055) is a new and highly selective HDAC inhibitor, which was approved by CFDA in China for the treatment of patients with refractory and relapsed PTCL as single agent. The synergistic effect of Chidamide based combination therapies has been rarely reported. In 2017, a report about Chidamide in combination with doxorubicin in PTCL cells has been published. In this study, the synergistic effect against PTCL cells exhibited after combination treatment with Chidamide and doxorubicin, inducing more significant apoptosis, changes of mitochondrial membrane potential and H3 acetylated level [161]. In a study of evaluation of the real-world utilization of Chidamide in 383 R/R PTCL patients in China, for 127 patients receiving Chidamide combined with chemotherapy, the therapeutic effects are impressive, with an ORR of 51.18%, DCR of 74.02%, PFS of 152 days (129 days for Chidamide as single agent) and an acceptable safety profile with more than 5% patients with thrombocytopenia (18.1%), neutropenia (12.6%), anemia (7.1%), and fatigue (5.5%) [98]. The therapeutic efficacy and safety profile of the combination of Chidamide with other targeted drugs or chemotherapy are favorable, offering an alternative choice for refractory and relapsed PTCL patients. However, the application of this combination treatment in clinic remains challenging, and further investigations are warranted.
3.2.5 Panobinostat guided combination therapies
HDAC inhibitors were reported to have synergistic antitumor activity with mTOR inhibitors based on their mechanisms of action. The combination of Panobinostat with Bortezomib exhibited highly synergistic efficacy against PTCL via inducing polyubiquitinated protein accumulation in preclinical studies, promoting its further exploration in clinical trials [162, 163]. In a phase I study of the combination of Panobinostat plus Everolimus in patients with relapsed or refractory Hodgkin and non-Hodgkin lymphoma (T-cell lymphomas included), highly synergistically therapeutic effects were demonstrated, 33% patients (indolent lymphoma, T-cell lymphoma, mantle cell lymphoma, and Hodgkin lymphoma) achieved objective responses and decreased multiple serum cytokine levels [164]. However, the adverse effects of this combination were most dose limiting severe toxicities of grade 3/4, including thrombocytopenia (64%), neutropenia (47%), anemia (20%), infection (10%), fatigue (7%), and dyspnea (7%). The promising therapeutic merits of the combination of Panobinostat with Everolimus offer new opportunities and improvements for the exploration of the Panobinostat based combination therapies against T-cell lymphomas in the future.
Paobinostat was demonstrated to have synergistic anti-lymphoma activity with proteasome inhibitor Bortezomib in many preclinical studies, advancing the clinical application in patients with relapsed or refractory peripheral T-cell lymphomas. An open-label, multicenter phase II trial of this combination was undertaken in patients with relapsed or refractory peripheral T-cell lymphoma showed that this combination therapy exhibited promising efficacy with acceptable safety and feasibility [137]. 43% patients displayed critical objective responses (including 20% complete responses). The common adverse effects of this combination therapy were grade 3/4, including thrombocytopenia (68%), neutropenia (40%), diarrhoea (20%), and asthenia or fatigue (8%), while 40% exhibited serious adverse events and 40% patients had peripheral neuropathy of any grade. The combination of Panobinostat with subcutaneous Bortezomib improved the outcomes (tolerance, treatment duration and safety profile) in patients with relapsed multiple myeloma in a randomized phase III non-inferiority study [165]. The combination of proteasome and HDAC inhibitors demonstrated encouraging therapeutic effects and acceptable safety profile in patients with PTCL, representing a feasible and adjustable therapeutic strategy for the treatment of patients with PTCL.
Besides, the studies of combination therapy of Panobinostat with Azacitidine in acute Myeloid Leukemia and Myelodysplastic Syndromes exhibited synergestic therapeutic effects and favorable safety profile[166], implying that Panobinostat may play an important role in the treatment of a range of hematologic malignancies.
All these findings validated the promising synergistic effect of Panobinostat in combination with other agents, which offer a novel therapeutic choice for T-cell lymphoma, especially peripheral T-cell lymphoma.
3.2.6 AN-7 guided combination therapy
Butyroyloxymethyl diethylphosphate (AN-7) is a novel HDAC inhibitor, showing selective anticancer activity in several cell lines (MF/SS cell lines and peripheral blood lymphocytes (PBL) from patients with Sézary syndrome (SPBL)) and animal models [167]. When combined with doxorubicin, more cell death was induced in both MF/SS cell lines and SPBL, but exhibited antagonistic effect in NPBL. The synergistic effect of AN-7 with doxorubicin in both MF/SS cell lines and SPBL offers promise for AN-7 as a novel HDAC inhibitor in the combinational therapy of T-cell lymphoma.
4. Perspectives
T-cell lymphomas are a heterogeneous group of lymphomas with complicate pathogenesis and poor prognosis. Novel therapeutic agents or strategies against T-cell lymphomas are urgently warranted, but with great challenges. HDACs are key epigenetic modulators important for the biological homeostasis. The specific functions and structure make them promising drug targets. A series of HDAC inhibitors with divergent characteristics have been developed and applied in the treatment of several malignancies, many of which displayed high potency against many T-cell lymphomas, especially HTLV-1-infected T-cells, peripheral T-cell lymphoma (PTCL), cutaneous T-cell lymphoma (CTCL), and acute T-cell lymphoma (ATL). Because of the limited efficacy, safety profile, and resistance, HDAC inhibitors approved as single therapeutic agents for T-cell lymphomas are rare. To date, only Vorinostat (SAHA) for CTCL, Romidepsin for CTCL and PTCL, and Belinostat for PTCL have been approved by the FDA, and Chidamide has been approved by the CFDA for the treatment of relapsed/refractory TCL as a single agent. The application of HDAC inhibitors in the treatment of T-cell lymphomas needs diligent optimization of the structure, potency, pharmacokinetics and safety. Combination therapy employing at least two pharmaceuticals or treatments possesses many advantages over single agent therapy, such as, improved therapeutic efficacy, abated adverse effects, resistance overcome, and more therapeutic strategy optimizations based on different action of modes (mechanisms). Within the past few decades, combination therapies have been the first choice of therapeutic strategies in oncology and other diseases with complicated pathogenesis and achieved remarkable curative effects. HDACs guided combination therapies have been used in the treatment of T-cell lymphomas. And many clinical trials have been undertaken in the treatment of T-cell lymphomas, exhibiting improved therapeutic merits and acceptable adverse effects in most combination regimens of HDAC inhibitors. Although anticipated therapeutic efficacy of HDAC guided combination therapies were demonstrated in these preclinical studies and clinical trials, extensive, randomized and comparative clinical trials are needed to further evaluate the most suitable combination regimens. Besides, according to the outcomes of reported combination therapies, there are still many aspects to further optimize, such as regimens in the combination (other targeted agents, such as checkpoint inhibitors Ipilimumab [3], small PARP inhibitor PJ-34 [168] or chemotherapies or other immune therapies (antibodies against CTLA4 and PD1/PDL1 [3], BiTE Blinatumomab [6])), dosage of each regimen, dosing optimizing and administration scheduling. The precise pathogenesis of T-cell lymphomas and thorough knowledge about HDACs would help to identify novel therapeutic targets. Gene sequencing, novel genetic editing techniques [169] and application of chemical probes in the elucidation of biomolecule functions [170] would help the understanding of drug targets in depth. De-novo drug developments or novel application of existed drugs would facilitate the flourish of HDAC inhibitors as single agent or combination regimen. Combination therapy designs should focus on optimizing the specific regimen, dosing, and administration schedule with the aim to dramatically improve synergistic therapeutic efficacy and minimize adverse effects. In all attempts to improve the therapeutic efficacy of T-cell lymphomas, the patients' status both physical (comorbidities and toxicity profile) and mental should be comprehensively evaluated. All in all, a bright future is on the way to defeat T-cell lymphomas after considering all these relevant elements and endeavor.
Abbreviations
Cutaneous T-cell lymphoma (CTCL), peripheral T-cell lymphoma (PTCL), acute T-cell lymphoma (ATL), Histone deacetylase (HDAC), nicotinamide adenine dinucleotide (NAD), tricostatin A (TSA), overall response rate (ORR), disease control rate (DCR), median progression-free survival (PFS), adverse events (AEs), the China Food and Drug Administration (CFDA), Europe European Medical Association (EMA), uridine diphosphate glucuronosyl-transferase (UGT), multiple myeloma (MM), indolent NHL, relapsed/refractory acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), Waldenstrom's Macroglobulinemia (WM), Polycomb repressive complex 2 (PRC2), Enhancer of zeste homolog 2 (EZH2), mycosis fungoides (MF).
Acknowledgements
This work is supported in part by grants from Sanming Projects of Medicine in Shenzhen (SZSM201612071), Shenzhen Foundation of Science and Technology (JCYJ20170306091928754, JCYJ20170816105345191), Shenzhen Foundation of Health and Family Planning Commission (SZBC2017028). We really appreciate the help of Prof. Dayue Fan for the language revision.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sorigue M, Junca J, Marce S, Cabezon M, Garcia O, Zamora L. The role of T-cell phenotype and T-cell receptor rearrangement in the diagnosis of T-cell malignancies. Leuk Lymphoma. 2016;57:244-6
2. Kato K, Akashi K. Recent Advances in Therapeutic Approaches for Adult T-cell Leukemia/Lymphoma. Viruses. 2015;7:6604-12
3. Matsuki E, Younes A. Checkpoint Inhibitors and Other Immune Therapies for Hodgkin and Non-Hodgkin Lymphoma. Curr Treat Options Oncol. 2016;17:31
4. Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC. et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem. 2007;2:58-61
5. Wu J, Fu J, Zhang M, Liu D. AFM13: a first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. Journal of hematology & oncology. 2015;8:96
6. Rader C. DARTs take aim at BiTEs. Blood. 2011;117:4403-4
7. Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, Zhang Q. et al. The Addition of the BTK Inhibitor Ibrutinib to Anti-CD19 Chimeric Antigen Receptor T Cells (CART19) Improves Responses against Mantle Cell Lymphoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:2684-96
8. Grupp SA. Advances in T-cell therapy for ALL. Best practice & research Clinical haematology. 2014;27:222-8
9. Cowell IG, Papageorgiou N, Padget K, Watters GP, Austin CA. Histone deacetylase inhibition redistributes topoisomerase IIbeta from heterochromatin to euchromatin. Nucleus. 2011;2:61-71
10. Zhang Q, Dai Y, Cai Z, Mou L. HDAC Inhibitors: Novel Immunosuppressants for Allo- and Xeno- Transplantation. ChemistrySelect. 2018;3:176-87
11. Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. 2016:6
12. Moskowitz AJ, Horwitz SM. Targeting histone deacetylases in T-cell lymphoma. Leukemia & lymphoma. 2017;58:1306-19
13. Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu C, Sakamoto KM. Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008;68:2557-60
14. Kekatpure VD, Dannenberg AJ, Subbaramaiah K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J Biol Chem. 2009;284:7436-45
15. Wang JC, Kafeel MI, Avezbakiyev B, Chen C, Sun Y, Rathnasabapathy C. et al. Histone deacetylase in chronic lymphocytic leukemia. Oncology. 2011;81:325-9
16. Zhang H, Shang YP, Chen HY, Li J. Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatol Res. 2017;47:149-59
17. Bodiford A, Bodge M, Talbott MS, Reddy NM. Profile of belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Onco Targets Ther. 2014;7:1971-7
18. Hood K, Shah A. Belinostat for Relapsed or Refractory Peripheral T-Cell Lymphoma. Journal of the advanced practitioner in oncology. 2016;7:209-18
19. Li Z, Zhu WG. Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications. Int J Biol Sci. 2014;10:757-70
20. Hagelkruys A, Sawicka A, Rennmayr M, Seiser C. The biology of HDAC in cancer: the nuclear and epigenetic components. Handbook of experimental pharmacology. 2011;206:13-37
21. Shen L, Orillion A, Pili R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics. 2016;8:415-28
22. Stengel KR, Zhao Y, Klus NJ, Kaiser JF, Gordy LE, Joyce S. et al. Histone Deacetylase 3 Is Required for Efficient T Cell Development. Molecular and cellular biology. 2015;35:3854-65
23. Kunami N, Katsuya H, Nogami R, Ishitsuka K, Tamura K. Promise of combining a Bcl-2 family inhibitor with bortezomib or SAHA for adult T-cell leukemia/lymphoma. Anticancer research. 2014;34:5287-94
24. Loosveld M, Castellano R, Gon S, Goubard A, Crouzet T, Pouyet L. et al. Therapeutic targeting of c-Myc in T-cell acute lymphoblastic leukemia, T-ALL. Oncotarget. 2014;5:3168-72
25. Palermo R, Checquolo S, Giovenco A, Grazioli P, Kumar V, Campese AF. et al. Acetylation controls Notch3 stability and function in T-cell leukemia. Oncogene. 2012;31:3807-17
26. Shao RH, Tian X, Gorgun G, Urbano AG, Foss FM. Arginine butyrate increases the cytotoxicity of DAB(389)IL-2 in leukemia and lymphoma cells by upregulation of IL-2Rbeta gene. Leukemia research. 2002;26:1077-83
27. Mishra A, La Perle K, Kwiatkowski S, Sullivan LA, Sams GH, Johns J. et al. Mechanism, Consequences, and Therapeutic Targeting of Abnormal IL15 Signaling in Cutaneous T-cell Lymphoma. Cancer discovery. 2016;6:986-1005
28. Troeger A, Glouchkova L, Ackermann B, Escherich G, Hanenberg H, Janka G. et al. Significantly increased CD70 up regulation on TEL-AML positive B cell precursor acute lymphoblastic leukemia cells following CD40 stimulation. Klinische Padiatrie. 2014;226:332-7
29. Fu W, Yi S, Qiu L, Sun J, Tu P, Wang Y. BCL11B-Mediated Epigenetic Repression Is a Crucial Target for Histone Deacetylase Inhibitors in Cutaneous T-Cell Lymphoma. The Journal of investigative dermatology. 2017
30. Ding H, Peterson KL, Correia C, Koh B, Schneider PA, Nowakowski GS. et al. Histone deacetylase inhibitors interrupt HSP90*RASGRP1 and HSP90*CRAF interactions to upregulate BIM and circumvent drug resistance in lymphoma cells. Leukemia. 2016
31. Lu X, Ning Z, Li Z, Cao H, Wang X. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis Res. 2016;5:185-91
32. Liu Z, Ding K, Li L, Liu H, Wang Y, Liu C. et al. A novel histone deacetylase inhibitor Chidamide induces G0/G1 arrest and apoptosis in myelodysplastic syndromes. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2016;83:1032-7
33. Rozati S, Cheng PF, Widmer DS, Fujii K, Levesque MP, Dummer R. Romidepsin and Azacitidine Synergize in their Epigenetic Modulatory Effects to Induce Apoptosis in CTCL. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:2020-31
34. Janik JE, Morris JC. Survivin(g) adult T-cell leukemia/lymphoma. Oncology. 2009;23:1256 61, 66
35. Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6561-5
36. Karelia N, Desai D, Hengst JA, Amin S, Rudrabhatla SV, Yun J. Selenium-containing analogs of SAHA induce cytotoxicity in lung cancer cells. Bioorganic & medicinal chemistry letters. 2010;20:6816-9
37. Grant S, Easley C, Kirkpatrick P. Vorinostat. Nature reviews Drug discovery. 2007;6:21-2
38. Molecule of the month. Vorinostat. Drug news & perspectives. 2006;19:352
39. Nishioka C, Ikezoe T, Yang J, Komatsu N, Bandobashi K, Taniguchi A. et al. Histone deacetylase inhibitors induce growth arrest and apoptosis of HTLV-1-infected T-cells via blockade of signaling by nuclear factor kappaB. Leuk Res. 2008;32:287-96
40. Zhang C, Richon V, Ni X, Talpur R, Duvic M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. The Journal of investigative dermatology. 2005;125:1045-52
41. Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C. et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109:31-9
42. Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S. et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2007;25:3109-15
43. Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert opinion on investigational drugs. 2007;16:1111-20
44. Richon VM, Garcia-Vargas J, Hardwick JS. Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett. 2009;280:201-10
45. Fraczek J, Vanhaecke T, Rogiers V. Toxicological and metabolic considerations for histone deacetylase inhibitors. Expert opinion on drug metabolism & toxicology. 2013;9:441-57
46. Kavanaugh SM, White LA, Kolesar JM. Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. Am J Health Syst Pharm. 2010;67:793-7
47. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. The oncologist. 2007;12:1247-52
48. Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals. 2010;3:2751-67
49. Duvic M, Dimopoulos M. The safety profile of vorinostat (suberoylanilide hydroxamic acid) in hematologic malignancies: A review of clinical studies. Cancer Treat Rev. 2016;43:58-66
50. Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG. et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060-6
51. Apuri S, Sokol L. An overview of investigational Histone deacetylase inhibitors (HDACis) for the treatment of non-Hodgkin's lymphoma. Expert opinion on investigational drugs. 2016;25:687-96
52. Ogura M, Ando K, Suzuki T, Ishizawa K, Oh SY, Itoh K. et al. A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. British journal of haematology. 2014;165:768-76
53. How J, Minden MD, Brian L, Chen EX, Brandwein J, Schuh AC. et al. A phase I trial of two sequence-specific schedules of decitabine and vorinostat in patients with acute myeloid leukemia. Leukemia & lymphoma. 2015;56:2793-802
54. Stahl M, Gore SD, Vey N, Prebet T. Lost in translation? Ten years of development of histone deacetylase inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Expert opinion on investigational drugs. 2016;25:307-17
55. Montalban-Bravo G, Huang X, Naqvi K, Jabbour E, Borthakur G, DiNardo CD. et al. A clinical trial for patients with acute myeloid leukemia or myelodysplastic syndromes not eligible for standard clinical trials. Leukemia. 2017
56. Singh MM, Johnson B, Venkatarayan A, Flores ER, Zhang J, Su X. et al. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro-oncology. 2015;17:1463-73
57. Diss E, Nalabothula N, Nguyen D, Chang E, Kwok Y, Carrier F. VorinostatSAHA Promotes Hyper-Radiosensitivity in Wild Type p53 Human Glioblastoma Cells. Journal of clinical oncology and research. 2014:2
58. Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM. et al. Vorinostat in solid and hematologic malignancies. J Hematol Oncol. 2009;2:31
59. Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T. et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. The Journal of antibiotics. 1994;47:301-10
60. Ueda H, Nakajima H, Hori Y, Goto T, Okuhara M. Action of FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum no. 968, on Ha-ras transformed NIH3T3 cells. Bioscience, biotechnology, and biochemistry. 1994;58:1579-83
61. Rajendran P, Williams DE, Ho E, Dashwood RH. Metabolism as a key to histone deacetylase inhibition. Crit Rev Biochem Mol Biol. 2011;46:181-99
62. VanderMolen KM, McCulloch W, Pearce CJ, Oberlies NH. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. The Journal of antibiotics. 2011;64:525-31
63. Imam MH, Shenoy PJ, Flowers CR, Phillips A, Lechowicz MJ. Incidence and survival patterns of cutaneous T-cell lymphomas in the United States. Leukemia & lymphoma. 2013;54:752-9
64. Reddy SA. Romidepsin for the treatment of relapsed/refractory cutaneous T-cell lymphoma (mycosis fungoides/Sezary syndrome): Use in a community setting. Crit Rev Oncol Hematol. 2016;106:99-107
65. Yu P, Petrus MN, Ju W, Zhang M, Conlon KC, Nakagawa M. et al. Augmented efficacy with the combination of blockade of the Notch-1 pathway, bortezomib and romidepsin in a murine MT-1 adult T-cell leukemia model. Leukemia. 2015;29:556-66
66. Deng C, Lipstein M, Rodriguez R, Serrano XO, McIntosh C, Tsai WY. et al. The novel IKK2 inhibitor LY2409881 potently synergizes with histone deacetylase inhibitors in preclinical models of lymphoma through the downregulation of NF-kappaB. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:134-45
67. Shustov A, Coiffier B, Horwitz S, Sokol L, Pro B, Wolfson J. et al. Romidepsin is effective and well tolerated in older patients with peripheral T-cell lymphoma: analysis of two phase II trials. Leukemia & lymphoma. 2017:1-7
68. Foss F, Pro B, Miles Prince H, Sokol L, Caballero D, Horwitz S. et al. Responses to romidepsin by line of therapy in patients with relapsed or refractory peripheral T-cell lymphoma. Cancer medicine. 2017;6:36-44
69. Chan KL, van der Weyden C, Khoo C, Lade S, Blombery P, Westerman D. et al. Durable clinical remission induced by romidepsin for chemotherapy-refractory peripheral T-cell lymphoma with central nervous system involvement. Leukemia & lymphoma. 2017;58:996-8
70. Irle C, Weintraub J. Long-Term Treatment with Romidepsin in Patients with Peripheral T-Cell Lymphoma. Case reports in hematology. 2016;2016:8175957
71. Barbarotta L, Hurley K. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. Journal of the advanced practitioner in oncology. 2015;6:22-36
72. Iyer SP, Foss FF. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. Oncologist. 2015;20:1084-91
73. Dupuis J, Morschhauser F, Ghesquières H, Tilly H, Casasnovas O, Thieblemont C. et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: a non-randomised, phase 1b/2 study. The Lancet Haematology. 2015;2:e160-e5
74. Jain S, Jirau-Serrano X, Zullo KM, Scotto L, Palermo CF, Sastra SA. et al. Preclinical pharmacologic evaluation of pralatrexate and romidepsin confirms potent synergy of the combination in a murine model of human T-cell lymphoma. Clin Cancer Res. 2015;21:2096-106
75. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature reviews Drug discovery. 2006;5:769-84
76. Steele NL, Plumb JA, Vidal L, Tjornelund J, Knoblauch P, Rasmussen A. et al. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:804-10
77. Chowdhury S, Howell GM, Teggart CA, Chowdhury A, Person JJ, Bowers DM. et al. Histone deacetylase inhibitor belinostat represses survivin expression through reactivation of transforming growth factor beta (TGFbeta) receptor II leading to cancer cell death. The Journal of biological chemistry. 2011;286:30937-48
78. Foss F, Advani R, Duvic M, Hymes KB, Intragumtornchai T, Lekhakula A. et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. British journal of haematology. 2015;168:811-9
79. Reimer P, Chawla S. Long-term complete remission with belinostat in a patient with chemotherapy refractory peripheral T-cell lymphoma. Journal of hematology & oncology. 2013;6:69
80. Campbell P, Thomas CM. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Journal of oncology pharmacy practice: official publication of the International Society of Oncology Pharmacy Practitioners. 2017;23:143-7
81. Lee HZ, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, Habtemariam BA. et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:2666-70
82. Poole RM. Belinostat: first global approval. Drugs. 2014;74:1543-54
83. Molife LR, de Bono JS. Belinostat: clinical applications in solid tumors and lymphoma. Expert opinion on investigational drugs. 2011;20:1723-32
84. Savickiene J, Treigyte G, Valiuliene G, Stirblyte I, Navakauskiene R. Epigenetic and molecular mechanisms underlying the antileukemic activity of the histone deacetylase inhibitor belinostat in human acute promyelocytic leukemia cells. Anti-cancer drugs. 2014;25:938-49
85. Bailey H, McPherson JP, Bailey EB, Werner TL, Gupta S, Batten J. et al. A phase I study to determine the pharmacokinetics and urinary excretion of belinostat and metabolites in patients with advanced solid tumors. Cancer chemotherapy and pharmacology. 2016;78:1059-71
86. Kirschbaum MH, Foon KA, Frankel P, Ruel C, Pulone B, Tuscano JM. et al. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a California Cancer Consortium Study. Leukemia & lymphoma. 2014;55:2301-4
87. Valiuliene G, Stirblyte I, Jasnauskaite M, Borutinskaite V, Navakauskiene R. Anti-leukemic effects of HDACi Belinostat and HMTi 3-Deazaneplanocin A on human acute promyelocytic leukemia cells. European journal of pharmacology. 2017;799:143-53
88. Cashen A, Juckett M, Jumonville A, Litzow M, Flynn PJ, Eckardt J. et al. Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Annals of hematology. 2012;91:33-8
89. Ong PS, Wang L, Chia DM, Seah JY, Kong LR, Thuya WL. et al. A novel combinatorial strategy using Seliciclib((R)) and Belinostat((R)) for eradication of non-small cell lung cancer via apoptosis induction and BID activation. Cancer letters. 2016;381:49-57
90. Kong LR, Tan TZ, Ong WR, Bi C, Huynh H, Lee SC. et al. Belinostat exerts anti-tumor cytotoxicity through the ubiquitin-proteasome pathway in lung squamous cell carcinoma. Molecular oncology. 2017
91. Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS. et al. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer chemotherapy and pharmacology. 2012;69:901-9
92. Gong K, Xie J, Yi H, Li W. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. The Biochemical journal. 2012;443:735-46
93. Zhao S, Guo J, Zhao Y, Fei C, Zheng Q, Li X. et al. Chidamide, a novel histone deacetylase inhibitor, inhibits the viability of MDS and AML cells by suppressing JAK2/STAT3 signaling. American journal of translational research. 2016;8:3169-78
94. Hasegawa H, Bissonnette RP, Gillings M, Sasaki D, Taniguchi H, Kitanosono H. et al. Induction of apoptosis by HBI-8000 in adult T-cell leukemia/lymphoma is associated with activation of Bim and NLRP3. Cancer Sci. 2016;107:1124-33
95. Shi Y, Dong M, Hong X, Zhang W, Feng J, Zhu J. et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Annals of oncology: official journal of the European Society for Medical Oncology. 2015;26:1766-71
96. Shi Y, Jia B, Xu W, Li W, Liu T, Liu P. et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: a multicenter real-world study in China. J Hematol Oncol. 2017;10:69
97. Lin SH, Wang BY, Lin CH, Chien PJ, Wu YF, Ko JL. et al. Chidamide alleviates TGF-beta-induced epithelial-mesenchymal transition in lung cancer cell lines. Molecular biology reports. 2016;43:687-95
98. Zhou Y, Pan DS, Shan S, Zhu JZ, Zhang K, Yue XP. et al. Non-toxic dose chidamide synergistically enhances platinum-induced DNA damage responses and apoptosis in Non-Small-Cell lung cancer cells. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2014;68:483-91
99. Liu L, Qiu S, Liu Y, Liu Z, Zheng Y, Su X. et al. Chidamide and 5-flurouracil show a synergistic antitumor effect on human colon cancer xenografts in nude mice. Neoplasma. 2016;63:193-200
100. Liu L, Chen B, Qin S, Li S, He X, Qiu S. et al. A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochemical and biophysical research communications. 2010;392:190-5
101. Wang H, Guo Y, Fu M, Liang X, Zhang X, Wang R. et al. Antitumor activity of Chidamide in hepatocellular carcinoma cell lines. Molecular medicine reports. 2012;5:1503-8
102. Zhao B, He T. Chidamide, a histone deacetylase inhibitor, functions as a tumor inhibitor by modulating the ratio of Bax/Bcl-2 and P21 in pancreatic cancer. Oncology reports. 2015;33:304-10
103. Qiao Z, Ren S, Li W, Wang X, He M, Guo Y. et al. Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells. Biochemical and biophysical research communications. 2013;434:95-101
104. He M, Qiao Z, Wang Y, Kuai Q, Li C, Wang Y. et al. Chidamide Inhibits Aerobic Metabolism to Induce Pancreatic Cancer Cell Growth Arrest by Promoting Mcl-1 Degradation. PLoS One. 2016;11:e0166896
105. Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer letters. 2009;280:233-41
106. Richardson PG, Moreau P, Laubach JP, Maglio ME, Lonial S, San-Miguel J. Deacetylase inhibitors as a novel modality in the treatment of multiple myeloma. Pharmacol Res. 2017;117:185-91
107. Sivaraj D, Green MM, Gasparetto C. Panobinostat for the management of multiple myeloma. Future oncology. 2017;13:477-88
108. Greig SL. Panobinostat: A Review in Relapsed or Refractory Multiple Myeloma. Targeted oncology. 2016;11:107-14
109. Gertz MA. Panobinostat in multiple myeloma. The Lancet Haematology. 2016;3:e552-e3
110. Gavriatopoulou M, Terpos E, Kastritis E, Dimopoulos MA. Current treatment options and investigational drugs for Waldenstrom's Macroglobulinemia. Expert opinion on investigational drugs. 2017;26:197-205
111. Morabito F, Voso MT, Hohaus S, Gentile M, Vigna E, Recchia AG. et al. Panobinostat for the treatment of acute myelogenous leukemia. Expert opinion on investigational drugs. 2016;25:1117-31
112. Abe F, Kitadate A, Ikeda S, Yamashita J, Nakanishi H, Takahashi N. et al. Histone deacetylase inhibitors inhibit metastasis by restoring a tumor suppressive microRNA-150 in advanced cutaneous T-cell lymphoma. Oncotarget. 2017;8:7572-85
113. Duvic M, Dummer R, Becker JC, Poulalhon N, Ortiz Romero P, Grazia Bernengo M. et al. Panobinostat activity in both bexarotene-exposed and -naive patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. European journal of cancer. 2013;49:386-94
114. Shao W, Growney JD, Feng Y, O'Connor G, Pu M, Zhu W. et al. Activity of deacetylase inhibitor panobinostat (LBH589) in cutaneous T-cell lymphoma models: Defining molecular mechanisms of resistance. Int J Cancer. 2010;127:2199-208
115. Ellis L, Pan Y, Smyth GK, George DJ, McCormack C, Williams-Truax R. et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:4500-10
116. Berardi S, Caivano A, Ria R, Nico B, Savino R, Terracciano R. et al. Four proteins governing overangiogenic endothelial cell phenotype in patients with multiple myeloma are plausible therapeutic targets. Oncogene. 2012;31:2258-69
117. Sasaki D, Imaizumi Y, Hasegawa H, Osaka A, Tsukasaki K, Choi YL. et al. Overexpression of Enhancer of zeste homolog 2 with trimethylation of lysine 27 on histone H3 in adult T-cell leukemia/lymphoma as a target for epigenetic therapy. Haematologica. 2011;96:712-9
118. Campbell GR, Bruckman RS, Chu YL, Spector SA. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. The Journal of biological chemistry. 2015;290:5028-40
119. Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A. et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733-43
120. Fiskus W, Buckley K, Rao R, Mandawat A, Yang Y, Joshi R. et al. Panobinostat treatment depletes EZH2 and DNMT1 levels and enhances decitabine mediated de-repression of JunB and loss of survival of human acute leukemia cells. Cancer biology & therapy. 2009;8:939-50
121. Cai Y, Yan X, Zhang G, Zhao W, Jiao S. The predictive value of ERCC1 and p53 for the effect of panobinostat and cisplatin combination treatment in NSCLC. Oncotarget. 2015;6:18997-9005
122. Gupta M, Ansell SM, Novak AJ, Kumar S, Kaufmann SH, Witzig TE. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood. 2009;114:2926-35
123. McCann SA, Story SK. Histone Deacetylase Inhibitors in Cutaneous T-cell Lymphoma. Journal of the Dermatology Nurses' Association. 2013;5:305-13
124. DeAngelo DJ, Spencer A, Bhalla KN, Prince HM, Fischer T, Kindler T. et al. Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia. 2013;27:1628-36
125. Tan D, Phipps C, Hwang WY, Tan SY, Yeap CH, Chan YH. et al. Panobinostat in combination with bortezomib in patients with relapsed or refractory peripheral T-cell lymphoma: an open-label, multicentre phase 2 trial. The Lancet Haematology. 2015;2:e326-33
126. Lucas DM, Davis ME, Parthun MR, Mone AP, Kitada S, Cunningham KD. et al. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia. 2004;18:1207-14
127. Gore L, Rothenberg ML, O'Bryant CL, Schultz MK, Sandler AB, Coffin D. et al. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:4517-25
128. Kummar S, Gutierrez M, Gardner ER, Donovan E, Hwang K, Chung EJ. et al. Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:5411-7
129. Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S. et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109:2781-90
130. Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. Journal of hematology & oncology. 2010;3:5
131. Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898-941
132. Slingerland M, Guchelaar HJ, Gelderblom H. Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors. Anti-cancer drugs. 2014;25:140-9
133. Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:3958-69
134. Zhan P, Wang X, Liu X, Suzuki T. Medicinal Chemistry Insights into Novel HDAC Inhibitors: An Updated Patent Review (2012-2016). Recent Pat Anticancer Drug Discov. 2017;12:16-34
135. Sawas A, Radeski D, O'Connor OA. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: a perspective review. Ther Adv Hematol. 2015;6:202-8
136. Reimer P. New developments in the treatment of peripheral T-cell lymphoma - role of Belinostat. Cancer Manag Res. 2015;7:145-51
137. Tan D, Phipps C, Hwang WYK, Tan SY, Yeap CH, Chan YH. et al. Panobinostat in combination with bortezomib in patients with relapsed or refractory peripheral T-cell lymphoma: an open-label, multicentre phase 2 trial. The Lancet Haematology. 2015;2:e326-e33
138. Belinostat (Beleodaq) for peripheral T-Cell lymphoma. The Medical letter on drugs and therapeutics. 2015; 57: e66-7.
139. Patrick B. Johnston AFC, Petros G. Nikolinakos, Anne W Beaven, Stefan Klaus Barta, Gajanan Bhat, Tao Song, Mi Rim Choi, Lee F. Allen, Sven de Vos, Yasuhiro Oki, Changchun Deng and Francine M Foss. Safe and effective treatment of patients with peripheral T-cell lymphoma (PTCL) with the novel HDAC inhibitor, belinostat, in combination with CHOP: Results of the bel-CHOP phase 1 trial. Blood. 2015;126:253
140. Nieto Y, Valdez BC, Thall PF, Jones RB, Wei W, Myers A. et al. Double epigenetic modulation of high-dose chemotherapy with azacitidine and vorinostat for patients with refractory or poor-risk relapsed lymphoma. Cancer. 2016;122:2680-8
141. Nieto Y, Valdez BC, Thall PF, Ahmed S, Jones RB, Hosing C. et al. Vorinostat Combined with High-Dose Gemcitabine, Busulfan, and Melphalan with Autologous Stem Cell Transplantation in Patients with Refractory Lymphomas. Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation. 2015;21:1914-20
142. Chen R, Frankel P, Popplewell L, Siddiqi T, Ruel N, Rotter A. et al. A phase II study of vorinostat and rituximab for treatment of newly diagnosed and relapsed/refractory indolent non-Hodgkin lymphoma. Haematologica. 2015;100:357-62
143. Oki Y, Younes A, Copeland A, Hagemeister F, Fayad LE, McLaughlin P. et al. Phase I study of vorinostat in combination with standard CHOP in patients with newly diagnosed peripheral T-cell lymphoma. British journal of haematology. 2013;162:138-41
144. Sanli H, Akay BN, Anadolu R, Ozcan M, Saral S, Akyol A. The efficacy of vorinostat in combination with interferon alpha and extracorporeal photopheresis in late stage mycosis fungoides and Sezary syndrome. Journal of drugs in dermatology: JDD. 2011;10:403-8
145. Wozniak MB, Villuendas R, Bischoff JR, Aparicio CB, Martinez Leal JF, de La Cueva P. et al. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica. 2010;95:613-21
146. Hutt DM, Roth DM, Vignaud H, Cullin C, Bouchecareilh M. The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PloS one. 2014;9:e106224
147. Dummer R, Beyer M, Hymes K, Epping MT, Bernards R, Steinhoff M. et al. Vorinostat combined with bexarotene for treatment of cutaneous T-cell lymphoma: in vitro and phase I clinical evidence supporting augmentation of retinoic acid receptor/retinoid X receptor activation by histone deacetylase inhibition. Leukemia & lymphoma. 2012;53:1501-8
148. Heider U, Rademacher J, Lamottke B, Mieth M, Moebs M, von Metzler I. et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. European journal of haematology. 2009;82:440-9
149. Hopfinger G, Nosslinger T, Lang A, Linkesch W, Melchardt T, Weiss L. et al. Lenalidomide in combination with vorinostat and dexamethasone for the treatment of relapsed/refractory peripheral T cell lymphoma (PTCL): report of a phase I/II trial. Annals of hematology. 2014;93:459-62
150. Sung JJ, Ververis K, Karagiannis TC. Histone deacetylase inhibitors potentiate photochemotherapy in cutaneous T-cell lymphoma MyLa cells. Journal of photochemistry and photobiology B, Biology. 2014;131:104-12
151. Dupuis J, Morschhauser F, Ghesquieres H, Tilly H, Casasnovas O, Thieblemont C. et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: a non-randomised, phase 1b/2 study. The Lancet Haematology. 2015;2:e160-5
152. Dai Chihara YO, Jason R Westin, Loretta Nastoupil, Luis E. Fayad, Felipe Samaniego, Emily Wesson, Charnelle Ruben, Sandra B. Horowitz, Lei Feng, Naveen Garg, Sairah Ahmed, Issa F. Khouri, Chitra M. Hosing, Jorge E. Romaguera, Nathan Fowler and Michelle A. Fanale. High Response Rate of Romidepsin in Combination with ICE (Ifosfamide, Carboplatin and Etoposide) in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma: Updates of Phase I Trial. Blood. 2015;126:3987
153. Cosenza M, Civallero M, Fiorcari S, Pozzi S, Marcheselli L, Bari A. et al. The histone deacetylase inhibitor romidepsin synergizes with lenalidomide and enhances tumor cell death in T-cell lymphoma cell lines. Cancer biology & therapy. 2016:1-13
154. Lunning MA, Ruan J, Nair S, Boruchov AM, Byrne R, Gerecitano JF. et al. A phase I/II trial of the combination of romidepsin and lenalidomide in patients with relapsed/refractory lymphoma and myeloma: Phase I results. Journal of Clinical Oncology. 2014
155. Kelly Zullo YG, Laurence Cooke, Xavier Jirau Serrano, Michael Mangone, Luigi Scotto, Jennifer E. Amengual, Daruka Mahadevan and Owen A. O'Connor. The investigational aurora A kinase inhibitor alisertib exhibits broad activity in preclinical models of T-cell lymphoma and is highly synergistic with romidepsin. Blood. 2014;124:4493
156. Michelle A. Fanale FBH, Luis Fayad, Yasuhiro Oki, Nathan Fowler, Jorge Romaguera, Nina Shah, Hubert Chuang, Lei Feng, Sandra B Horowitz, Emily Wesson, Toni Y Hutto, Tariq Muzzafar, Felipe Samaniego and R Eric Davis. A Phase I Trial of Alisertib Plus Romidepsin for Relapsed/Refractory Aggressive B- and T-Cell Lymphomas. Blood; 2014. p. 1744
157. Pellegrini C, Dodero A, Chiappella A, Monaco F, Degl'Innocenti D, Salvi F. et al. A phase II study on the role of gemcitabine plus romidepsin (GEMRO regimen) in the treatment of relapsed/refractory peripheral T-cell lymphoma patients. Journal of hematology & oncology. 2016;9:38
158. Chakraborty AR, Robey RW, Luchenko VL, Zhan Z, Piekarz RL, Gillet JP. et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: rationale to combine romidepsin with an MEK inhibitor. Blood. 2013;121:4115-25
159. Akilov OE, Grant C, Frye R, Bates S, Piekarz R, Geskin LJ. Low-dose electron beam radiation and romidepsin therapy for symptomatic cutaneous T-cell lymphoma lesions. The British journal of dermatology. 2012;167:194-7
160. Samimi S, Morrissey K, Anshelevich S, Evans K, Gardner J, Musiek A. et al. Romidepsin and interferon gamma: a novel combination for refractory cutaneous T-cell lymphoma. Journal of the American Academy of Dermatology. 2013;68:e5-6
161. Zhang H, Dong L, Chen Q, Kong L, Meng B, Wang H. et al. Synergistic antitumor effect of histone deacetylase inhibitor and Doxorubicin in peripheral T-cell lymphoma. Leuk Res. 2017;56:29-35
162. Phipps C, Mok SJ, Ang AL, Goh YT, Tan D. Bortezomib and panobinostat combination is effective against PTCL. Leuk Res. 2012;36:e128-30
163. Ocio EM, Vilanova D, Atadja P, Maiso P, Crusoe E, Fernandez-Lazaro D. et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica. 2010;95:794-803
164. Oki Y, Buglio D, Fanale M, Fayad L, Copeland A, Romaguera J. et al. Phase I study of panobinostat plus everolimus in patients with relapsed or refractory lymphoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:6882-90
165. Arnulf B, Pylypenko H, Grosicki S, Karamanesht I, Leleu X, van de Velde H. et al. Updated survival analysis of a randomized phase III study of subcutaneous versus intravenous bortezomib in patients with relapsed multiple myeloma. Haematologica. 2012;97:1925-8
166. Khot A, Dickinson M, Prince HM. Panobinostat in lymphoid and myeloid malignancies. Expert opinion on investigational drugs. 2013;22:1211-23
167. Moyal L, Feldbaum N, Goldfeiz N, Rephaeli A, Nudelman A, Weitman M. et al. The Therapeutic Potential of AN-7, a Novel Histone Deacetylase Inhibitor, for Treatment of Mycosis Fungoides/Sezary Syndrome Alone or with Doxorubicin. PloS one. 2016;11:e0146115
168. Bai XT, Moles R, Chaib-Mezrag H, Nicot C. Small PARP inhibitor PJ-34 induces cell cycle arrest and apoptosis of adult T-cell leukemia cells. J Hematol Oncol. 2015;8:117
169. Valente LJ, Aubrey BJ, Herold MJ, Kelly GL, Happo L, Scott CL. et al. Therapeutic Response to Non-genotoxic Activation of p53 by Nutlin3a Is Driven by PUMA-Mediated Apoptosis in Lymphoma Cells. Cell reports. 2016;14:1858-66
170. Zhang Q, Liu H, Pan Z. A general approach for the development of fluorogenic probes suitable for no-wash imaging of kinases in live cells. Chemical communications. 2014;50:15319-22
Author contact
![]() Corresponding authors: Dr. Qing Zhang, email: zhangqing7864com; Dr. Zhendong Yu, email: dongboyaacom
Corresponding authors: Dr. Qing Zhang, email: zhangqing7864com; Dr. Zhendong Yu, email: dongboyaacom