Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(1):180-188. doi:10.7150/ijms.27719 This issue Cite
Research Paper
Nonselective Cyclooxygenase Inhibition Retards Cyst Progression in a Murine Model of Autosomal Dominant Polycystic Kidney Disease
Min Zhang1, Manakan B. Srichai2,4, Min Zhao2, Jian Chen2, Linda S. Davis2, Guanqing Wu2,3, Matthew D. Breyer5, Chuan-Ming Hao1,2,4 ![]()
1. Division of Nephrology, Huashan Hospital, Fudan University, Shanghai, China
2. Department of Medicine, Division of Nephrology, Vanderbilt University, Nashville, TN
3. Department of Cell and Developmental Biology, Vanderbilt University, Nashville, TN
4. VA Medical Center, Nashville, TN.
5. Biotechnology Discovery Research, Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, Indiana 46225, USA
Received 2018-6-7; Accepted 2018-12-7; Published 2019-1-1
Abstract
Aim: Autosomal dominant polycystic kidney disease is one of the most common genetic renal diseases. Cyclooxygenase plays an important role in epithelial cell proliferation and may contribute to the mechanisms underlying cyst formation. The aim of the present study was to evaluate the role of cyclooxygenase inhibition in the cyst progression in polycystic kidney disease.
Method: Pkd2WS25/- mice, a murine model which harbors a compound cis-heterozygous mutation of the Pkd2 gene were used. Cyclooxygenase expression was assessed in both human and murine kidney specimens. Pkd2WS25/- mice were treated with Sulindac (a nonselective cyclooxygenase inhibitor) or vehicle for 8 months starting at three weeks age, and then renal cyst burden was assessed by kidney weight and volume.
Results: Cyclooxygenase-2 expression was up-regulated compared to control kidneys as shown by RNase protection in human polycystic kidneys and immunoblot in mouse Pkd2WS25/- kidneys. Cyclooxygenase-2 expression was up-regulated in the renal interstitium as well as focal areas of the cystic epithelium (p<0.05). Basal Cyclooxygenase-1 levels were unchanged in both immunohistochemistry and real-time PCR. Administration of Sulindac to Pkd2WS25/- mice and to control mice for 8 months resulted in reduced kidney weights and volume in cystic mice. Renal function and electrolytes were not significantly different between groups.
Conclusion: Thus treatment of a murine model of polycystic kidney disease with Sulindac results in decreased kidney cyst burden. These findings provide additional implications for the use of Cyclooxygenase inhibition as treatment to slow the progression of cyst burden in patients with polycystic kidney disease.
Keywords: ADPKD, COX2, PKD2, prostanoid, Sulindac
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common genetic diseases, occurring in 1:400 to 1:1000 live births. In the U.S. ADPKD accounts for an estimated 4.6% of patients with end stage renal disease (ESRD) with an annual ESRD incidence rate of about 6.9 per million [1]. Despite the ability to diagnose ADPKD early in life, therapeutic options to forestall the predestined progression to renal failure remain limited. By the age of 65 years, about 45% to 70% of affected individuals have developed ESRD [2]. Thus further investigations into new therapies are critical to further impact the prognosis of ADPKD. Several recent advances including the availability of animal models which mimic polycystic disease in humans have allowed a better understanding of the pathological mechanisms leading towards the progression of polycystic kidney disease [3]. These advances have led to focus therapeutic efforts targeted to the molecular basis of cyst formation and to the development of strategies aimed to reduce the rate of cyst expansion.
Mutations in either the PKD1 or PKD2 gene account for 85-90% and 10-15% of cases of ADPKD, respectively [4]. ESRD develops as a consequence of altered renal parenchyma resulting from the progressive expansion of epithelial cysts and subsequent remodeling of normal renal tissue interposed between expanding cysts [5]. Strong evidence also suggests that the progression of ADPKD is affected by multiple genetic and non-genetic modifiers, which is associated with substantial intra-familial phenotypic variations of ADPKD [6]. The identities of these modifiers and the mechanism by which these modifiers affect polycystic disease progress have not been completely defined.
Cyclooxygenase (COX) is a key enzyme involved in prostanoid biosynthesis which converts arachidonate to prostaglandin H2 (PGH2) which is then converted to 5 major bioactive prostanoids via distinct synthases. In the kidney, the two most abundant prostanoids are prostaglandin E2 (PGE2) and prostacyclin (PGI2), synthesized by microsomal PGE synthase (mPGES) and PGI synthase (PGIS), respectively [7]. COX exists as two major isoforms, COX1 and COX2. COX2 represents an inducible early growth response gene product that is up-regulated as cells shift from quiescence (Go) to proliferation (G1) [8]. COX derived prostanoids have been reported to promote cell proliferation and contribute to the development of intestinal polyposis in both mice and humans [9]. Inhibition of COX2 either by selective or non-selective inhibitors have been shown to markedly reduce intestinal polyps in mice and in humans [10]. In the kidney, accelerated renal epithelial proliferation has been uniformly demonstrated in ADPKD patients and in animal models, with formation of micro-polyps on the wall of kidney cysts [11]. Furthermore, elevated COX2 enzyme activity has been reported in a rat model of PKD [12]. Based on the similarities in cyst formation between intestinal polyposis and polycystic disease, we hypothesized that COX2 may be an important mediator in promoting epithelial cell proliferation and cyst expansion in polycystic kidney disease and that cyclooxygenase inhibitors may slow cyst progression. This hypothesis is supported by a recent study showing that COX2 inhibition is associated with reduced kidney cyst size in the Han: SPRD-cy rat, a rat model of autosomal dominant polycystic kidney disease [12]. Although the phenotype of the Han: SPRD rat resembles human ADPKD in many aspects, the gene responsible for development of PKD does not share homology relationships with PKD1 or PKD2 [13]. The present studies examined the role of COX2 in a compound cis-heterozygous murine model of human PKD2, Pkd2WS25/- [14]. This model has targeted mutations to the mouse homolog, Pkd2, which results in an unstable allele, WS25, that can undergo homologous recombination-based somatic rearrangements to form a null allele [14].
Methods
Experimental animals and treatment. All animal experiments were performed according to animal protocols approved by the Animal Care Use Committee at Vanderbilt University School of Medicine. Pkd2+/- mice were intercrossed with either Pkd2+/WS25 or Pkd2WS25/WS25 animals (kindly provided by Drs. Stefan Somlo and Guanqing Wu) to obtain compound cis-heterozygous Pkd2WS25/- mice [14]. Pkd2+/WS25 and Pkd2+/+ mice served as controls. All mice were housed in regular cages under normal 12hr light/dark cycles and were fed a standard rodent diet. At sacrifice, kidneys were harvested and snap frozen. Kidneys were also cut and fixed in 4% paraformaldehyde for paraffin sectioning. Blood urea nitrogen (BUN), hematocrit, and serum electrolytes were measured by a commercially available assay (Heska-iSTAT).
Pkd2WS25/- mice were treated with Sulindac (12 to 14 mg/kg body weight/day) [15, 16] or vehicle for 8 months starting at three weeks age. Sulindac was added to mouse chow (0.006%) and stored at 4°C. At this concentration, plasma concentration of Sulindac sulfide (the active component of Sulindac) was estimated to be 〜 0.5μg/ml [15]. For Sulindac sulfide, this concentration resulted in 〜50% inhibition of COX-1 and <50% inhibition of COX-2 in human whole blood assays [17]. Fresh food was prepared monthly. At the end of the study, renal cyst burden was assessed by kidney weight and kidney volume which was calculated by the standard formula for a modified ellipsoid (4/3π x (anteroposterior diameter/4 + width/4)2 x length/2).
Renal volume =
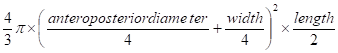
Human kidney tissue. Kidney nephrectomy specimens from adult ADPKD and control patients undergoing nephrectomy were obtained from Vanderbilt University surgical pathology with IRB approval. Total RNA and paraffin-embedded kidney sections were prepared.
FITC-inulin measurement of GFR. Glomerular filtration rate (GFR) was assessed according to clearance kinetics of plasma FITC-inulin following a single bolus injection. Briefly, 5% FITC-inulin was prepared by dissolving FITC-insulin (Sigma) in 0.9% NaCl. Dialyzed FITC-inulin was injected retro-orbitally (3.74μl/g body weight) under light anesthesia induced using Isoflurane (Baxter Pharmaceutical Products Inc., Deerfield, IL). Approximately 20μl of blood was collected via saphenous vein at 3, 7, 10, 15, 35, 55, 75 minutes post FITC-inulin injection. 10μl of plasma was used for FITC determination using a Fluoroscan Ascent FL (Labsystems, FIN-00811 Helsinki, Finland), with 485 nm excitation, and read at 538 nm emission. GFR was calculated according to two compartment clearance models using the formula: GFR = I/(A/a + B/b), where I is the amount of FITC-inulin delivered by the bolus injection; A and B are the y -intercept values of the two decay rates, and a and b are the decay constants for the distribution and elimination phases, respectively.
Western blotting. Whole kidney lysate was prepared by homogenizing kidneys in RIPA buffer (1% Triton X-100, 0.5% deoxycholate, 1% SDS, 150mM NaCl, 50mM Tris-Hcl (pH 8), 2mM EDTA, and 1mM sodium orthovanadate) with protease cocktail inhibitors (Complete Mini, Roche Diagnostics) using a homogenizer (PowerGen 700, Fisher Scientific). Samples were incubated on ice, passed through a 25g syringe, and centrifuged at 4°C for 30min 10000g. Supernatant was collected and protein levels measured (BCA, Sigma). Protein was electrophoresed on a 10% Tris-Hcl SDS gel (BioRad) and transferred to nitrocellulose membrane overnight. Membranes were blocked in blocking buffer (150 mM NaCl, 50 mM Tris, 0.05% Tween 20, and 5% Carnation nonfat dry milk, pH 7.5) for 1hr at room temperature, and incubated with affinity-purified primary COX2 Ab (1:1000, 160126 Cayman) or β-actin (1:10000 Sigma) in blocking buffer overnight at 4°C. After washing three times in TBST (50 mM Tris, pH 7.5, 150 mM NaCl, 0.05% Tween 20), the membranes were incubated with horseradish peroxidase-conjugated secondary antibody (1:5000 for anti-mouse and 1:10000 for anti-rabbit) for 1hr at room temperature, followed by three 15-min washings. Antibody labeling was visualized by addition of chemiluminescence reagent (Renaissance, DuPont-New England Nuclear, Boston, MA), and the membrane was exposed to Kodak XAR-5 film and developed.
Immunohistochemistry: Paraffin kidney sections were prepared and stained for COX2 1:1000 (Cayman, 160106) and mPGES1 (Cayman, 160140) using standard protocol. In general at the termination of an experiment, mice were deeply anesthetized with Nembutal (70mg/kg ip) and the right kidney was clamped and removed for immunoblot. Mice were then exsanguinated with 50mL/100g heparinized saline (0.9% NaCl, 2U/mL heparin, 0.02% sodium nitrite) through a transcardial aortic cannula and the left kidney fixed by retrograde perfusion through the heart with 4% paraformaldehyde. Tissue blocks were post-fixed overnight in 4% paraformaldehyde and subsequently dehydrated through a graded series of ethanol, embedded in paraffin, sectioned (4μm), and mounted on glass slides. Internal controls and comparisons were facilitated by creating compound blocks with multiple specimens that were sectioned and stained together. COX2 was immunolocalized with affinity-purified rabbit polyclonal anti-murine COX2 peptide (residues 570-598) (Cayman Chemicals, 160106) at a dilution of 1:1000. Staining was localized using a biotinylated anti-rabbit secondary antibody. Biotin was identified using streptavidin coupled to horseradish peroxidase and visualized with diaminobenzidine (Vector Vectastain ABC kit). Sections were viewed and imaged with a Zeiss Axioskop and Spot-Cam digital camera (Diagnostic Instruments).
In-situ hybridization. In-situ hybridization was performed as previously described [18]. Briefly, a 35S-labeled antisense riboprobe generated from the 597-bp PCR fragment of mouse or 471-bp PCR fragment of human 3' untranslated region of the COX2 cDNA was hybridized to the tissue sections and then washed as previously described. The slides were dehydrated with graded ethanol containing 300 mM ammonium acetate, dipped in emulsion (Ilford K5; Knutsford, Cheshire, UK) and exposed for 4-5 days at 4°C. After developing in Kodak D-19, the slides were counterstained with hematoxylin. Photomicrographs were taken using a Zeiss Axioskop microscope with either dark field or bright field optics.
Nuclease protection. A 471-bp riboprobe for nuclease protection was generated from a portion of the 3'region of the human COX2 cDNA. A 250-bp riboprobe was generated from a PCR fragment of human β-actin. Antisense cRNA was transcribed in the presence of [α-32P]-UTP 400-800 Ci/mmol (MAXIscript; Ambion, Austin, Texas, USA) and hybridized to 30 μg of total cellular RNA at 42°C for 14-18 hours. In all cases total cellular RNA was prepared using chloroform/phenol extraction using commercial reagents (Molecular Research Center Inc., Cincinnati, Ohio, USA). Ribonuclease A digestion was carried out at 37°C for 30 minutes. Protected fragments were separated on a 6% polyacrylamide/7 M urea gel, followed by autoradiography.
Statistical analysis. All statistical analyses were performed with Prism Version 4.0a for Macintosh. One-way ANOVA were performed for total cyst burden comparison.
Results
COX2 expression is up-regulated in polycystic kidneys from ADPKD patients
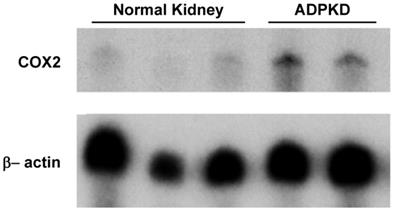
To assess COX2 expression levels in ADPKD kidneys, nuclease protection assay was performed. As shown in Figure 1, COX2 mRNA expression in diseased ADPKD kidneys was substantially up-regulated (Lanes 4-5) compared to normal, non-cystic kidneys (Lanes 1-3).
Nuclease protection assay for COX2 transcript expression. Lanes 1-3 represent 3 separate normal control adult human kidneys. Lanes 4-5 represent 2 separate adult ADPKD human kidneys. 30μg total RNA loaded per lane.
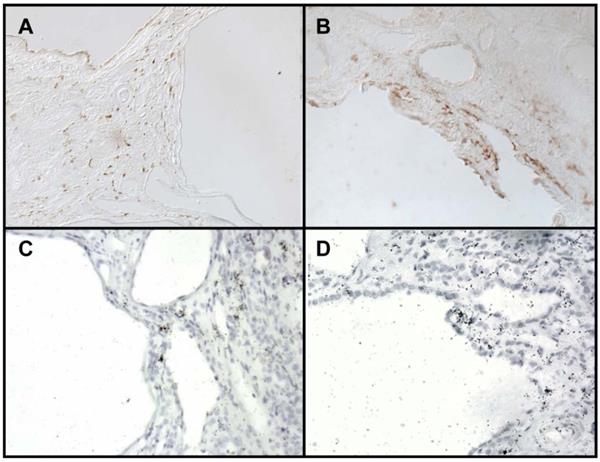
Immunohistochemistry for COX2 localized COX2 immunoreactivity to the renal interstitium as well as to the epithelial cells lining the cysts (Figure 2A, 2B). Localization to these areas was confirmed by in situ hybridization (Figure 2C, 2D).
COX2 protein localization in kidneys from ADPKD patients. (A-B) Immunohistochemistry reveals COX2 reactivity in the interstitium (A) and epithelial cyst lining (B). (C-D) In-situ hybridization for COX2 mRNA transcript. 200X brightfield.
COX and prostanoid synthase expression in cystic kidney of Pkd2 mice
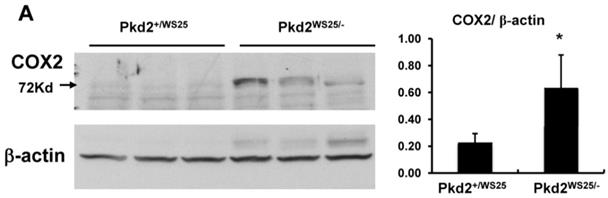
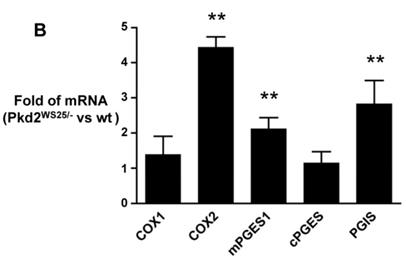
Kidney COX expression in a mouse model of human PKD2, Pkd2WS25/-, was examined via immunoblot and real-time PCR. Cystic kidneys from Pkd2WS25/- mice had increased COX2 immunoreactivity compared to non-cystic kidneys from Pkd2+/WS25 mice (Figure 3A, p<0.05). This difference in COX2 expression level was also noted at the mRNA transcript level by real-time PCR, whereas no differences were observed in COX1 (Figure 3B). In addition, the prostaglandin synthases, mPGES1 and PGIS, were elevated in Pkd2WS25/- mice by real-time PCR (Figure 3B) (n=4, p<0.01); no differences were observed in cytosolic PGE synthase (cPGES).
(A) Immunoblot for COX2 and β-actin in Pkd2+/WS25 and Pkd2WS25/- kidneys. 30μg protein lysate loaded per lane. * p<0.05. (B) Real-time PCR for COX and downstream prostanoid synthases in Pkd2WS25/- kidneys vs. control kidneys. N=4, ** p<0.01.
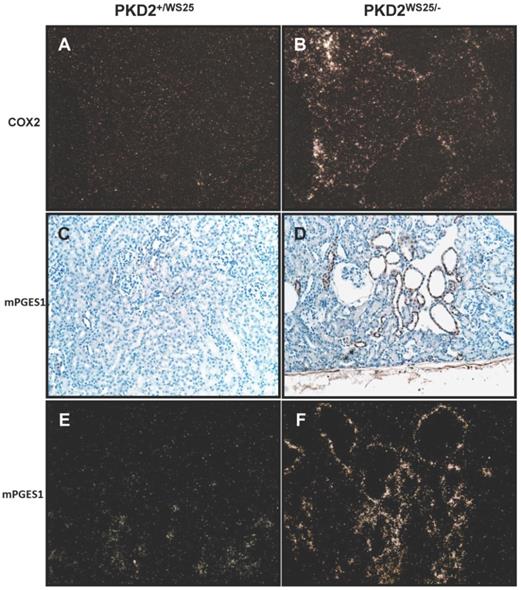
Localization of COX2 mRNA transcript by in situ hybridization demonstrated expression in cystic epithelial cells and in the interstitium, similar to the findings in human ADPKD kidneys (Figure 4B). Control kidneys had nearly absent COX2 mRNA expression (Figure 4A). Localization of mPGES1 by immunohistochemistry and in situ hybridization demonstrated expression primarily along the cystic epithelia cells (Figure 4C-F). Taken together, these studies suggest COX2 and the downstream prostanoids PGE2 and PGI2 may be associated with cyst development in Pkd2WS25/- mice.
Localization of COX2 and mPGES1 mRNA transcript in control Pkd2+/WS25 kidneys and cystic Pkd2WS25/- kidneys. (In-situ hybridization) (A-B) COX2 mRNA transcript in control Pkd2+/WS25 kidneys (A) and cystic Pkd2WS25/- kidneys (B). mPGES1 transcript in control Pkd2+/WS25 kidneys (C,E) and cystic Pkd2WS25/- (D,F) kidneys. Immunohistochemistry (C,D), In-situ hybridization (E,F). 200X darkfield (A-B, E-F); 200x brightfield (C-D).
Effect of cyclooxygenase inhibitor on cyst development and renal function
To determine the effects of COX inhibition on the progression of cyst growth in polycystic kidney disease, Pkd2WS25/- cystic mice were treated with Sulindac, a nonselective oral COX inhibitor. Kidney volumes of Pkd2WS25/- mice (1.19±0.83 cm3, n=9, 4 males) were significantly larger than that of control Pkd2+/WS25 and PKD2+/+ mice (0.54±0.07 cm3, n=21, 10 males, p<0.01, Figure 5A). Sulindac treatment significantly reduced the kidney volumes of Pkd2WS25/- mice (0.74±0.29 cm3, n=8, 3 males) when compared to vehicle-treated Pkd2WS25/- mice (p<0.05). Similar results were obtained when renal cyst burden was assessed by kidney weight (Figure 5B).
Combined kidney volumes (A) and weights (B) following 8mo treatment with vehicle or Sulindac. WT (n=21) represents kidneys from either Pkd2+/WS25 or Pkd2+/+ mice. (A) Kidney volume in vehicle-treated Pkd2WS25/- mice (n=9) were significantly elevated when compared to WT kidneys (** p<0.01) and when compared to Sulindac-treated Pkd2WS25/- mice (n=8) (# p<0.05). Combined kidney volumes of WT, Vehicle, Sulindac: 0.54±0.07, 1.19±0.83, 0.74±0.29. (B) Kidney weights were significantly higher in vehicle-treated Pkd2WS25/- kidneys when compared to WT kidneys (* p<0.01). Sulindac-treated Pkd2WS25/- mice had a trend towards lower weights compared to vehicle-treated Pkd2WS25/- although this did not approach statistical significance. Again there were no statistical differences in kidney weights between WT kidneys and Sulindac-treated Pkd2WS25/- kidneys.
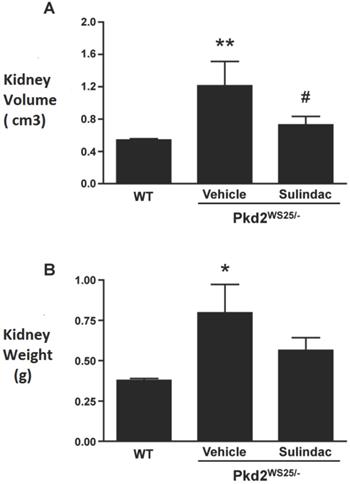
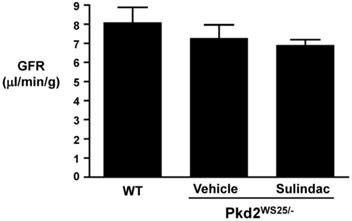
Table 1 shows the results of whole blood electrolytes and BUN as measured by i-STAT. There were no significant differences in BUN, hematocrit, or electrolytes between the three groups (Table 1). FITC-inulin clearance performed at the end of treatment revealed a slight decrease in GFR in Pkd2WS25/- mice as compared to control mice, however, these differences were not statistically significant (Figure 6).
Blood electrolyte and chemical data of control Pkd2+/WS25 kidneys and cystic Pkd2WS25/- kidneys treated with vehicle or Sulindac. No statistically different was observed.
| control | PKD2+V | PKD2+SU | |
|---|---|---|---|
| Na (mEq/L) | 150±1.9 | 150±2.9 | 151±6.4 |
| K (mEq/L) | 4.2±0.4 | 4.6±0.8 | 4.9±0.8 |
| Cl (mEq/L) | 119±4.1 | 123±5.0 | 121±6.6 |
| Glu (mEq/L) | 185±48 | 180±35 | 225±94 |
| pH | 7.30±0.03 | 7.31±0.07 | 7.30±0.04 |
| TCO2 | 22±2.9 | 23±4.2 | 24±2.4 |
| BUN (mg/dl) | 21±3.9 | 26±5.8 | 26±7.2 |
| Hct (%) | 34±3.6 | 28±3.6 | 30±2.5 |
PKD2+V: PKD2WS25/-+vehicle; PKD2+SU: PKD2WS25/-+Sulindac
GFR as assessed by FITC-inulin clearance. GFR in WT (8±1.60 l/min/g, n=4), vehicle-treated Pkd2WS25/- (7.26±1.22 l/min/g, n=3), and Sulindac-treated Pkd2WS25/- (6.89±0.52 l/min/g, n=3) mice were not statistically different.
Discussion
Renal cystic diseases represent an important cause of renal insufficiency and renal failure [19, 20]. Despite major advances in understanding the genetic basis for disease [14, 21, 22], the pathogenesis of cyst formation and expansion is incompletely understood. Furthermore, therapeutic medical interventions that can block kidney cyst expansion are currently limited. Animal studies have shown potential therapeutic roles for vasopressin V2 receptor antagonists and mTOR inhibitors in retarding cyst growth [23, 24]. More recently, the COX2 inhibitor, NS-398, was shown to reduce cyst growth in the Han: SPRD rats [12]. In the current study, we have examined the role of cyclooxygenase in human ADPKD and in a murine model of human PKD2, Pkd2WS25/-. We show increased COX2 expression in cystic kidneys from both human ADPKD and Pkd2WS25/- mice as well as increased expression of the downstream prostanoid synthases, mPGES1 and PGIS, in Pkd2WS25/- kidneys. Treatment with Sulindac, a nonselective COX inhibitor, significantly retarded increases in kidney volume in Pkd2WS25/- mice. Taken together, these data suggest that COX-derived prostanoids may represent potential therapeutic targets for ADPKD.
COX-derived prostaglandins have been documented to play important roles in many physiological and pathological processes, such as maintenance of fluid homeostasis and vascular tone, inflammation, angiogenesis, and cell proliferation. Non-selective COX inhibitors and COX2 inhibitors have been shown to inhibit cell proliferation and reduce the size and number of intestinal polyps in both humans with familial adenomoatous polyposis (FAP) and in animal models which mimic the disease. Accelerated cell proliferation has invariably been detected in renal cystic epithelial cells, and hyperplastic polyps are commonly observed within the epithelia lining the cyst wall [11]. A two-hit mechanism of cyst formation has been proposed which may account for the focal nature of cyst development [25]. The Pkd2WS25/- murine model of ADPKD supports the two-hit hypothesis, which was originally proposed by Knudsen to explain childhood tumors.
As with ADPKD, FAP also represents an autosomal dominant disease associated with benign (at least initially) but accelerated epithelial proliferation of intestinal cells. Two groups identified the relevant mutation in a novel gene designated APC (adenomatous polyposis coli). Polyps in FAP also develop sporadically as a result of loss of heterozygosity (LOH) due to somatic cell mutations in the remaining normal somatic cell APC allele. Furthermore there is evidence that the signaling pathways activated by the APC protein and polycystin-1 may be similar as both proteins may interact with the β-catenin-nuclear signaling pathway, altering gene expression and activating cell proliferation. These considerations provide a conceptual basis for utilizing similar approaches for the treatment of FAP and ADPKD. This hypothesis is supported by the present study which revealed increased COX2 expression in cystic kidneys of both human ADPKD and Pkd2WS25/- mice and reduced kidney volume in cystic mice following COX inhibition. We chose to study cyst growth in a murine model of PKD, Pkd2WS25/- because unlike other rodent models, this one more closely mimics human PKD with the second hit model. In this murine model, bilateral cyst formation occurs in 100% of animals by 10-11 wk age [14]. As in human ADPKD, formation of kidney cysts in this model is associated with renal failure and early death [14]. But to our experience and according to literatures, the decline of renal function in this mouse line is variable [14, 26]. Some studies shown Pkd2WS25/- mice had no significant BUN increase even at the age of 5 months [26]. Longitudinal FISP-MRI analyses of changes in cyst volumes were performed in this model over 15 months. The result shown that Pkd2WS25/- mice had significant increases in kidney weights at 4 months of age. Interestingly, between 4 and 12 months of age, there was no accelerated progression of kidney growth [27]. We chose to evaluate the effect of COX inhibitor in this model for 8 months in order to exclude the variety of renal function decline.
To examine whether COX derived prostanoids play a role in cyst growth in Pkd2 mice, Pkd2WS25/- mice were treated with Sulindac, which has been reported to significantly inhibit colorectal adenomatous polys in both rodent models and in patients with FAP [28-30]. Celecoxib (selective COX2 inhibitor) use resulted in a modest reduction of colorectal polyps, but is no longer US FDA-approved for this indication, due to lack of complete follow-up studies. Treatment was started at 3 weeks age to avoid renal dysgenesis caused by COX2 inhibition [31]. Sulindac treatment resulted in a significant reduction in kidney volume and did not significantly alter BUN or GFR levels. However, none of the polycystic animals, either treated or untreated, developed evidence for ESRD, likely because the disease is slowly progressive and renal function tends to deteriorate at later stages of the disease.
In ADPKD, changes in cyst volume occur prior to renal dysfunction. Cyst development and time-dependent structural disruption of renal architecture plays a central role in the genesis of PKD and cyst growth is a key predictor of deterioration of renal function. Despite the lack of an effect on renal function, the change of kidney volume is likely to have long-term beneficial effects on disease progression.
Furthermore, a concern raised regarding the use of NSAIDs is their potential to cause renal damage in patients with renal insufficiency. In this study, no significant difference of BUN was observed between vehicle treated group and Sulindac treated group. These results show the need for further investigation into the longer-term effects and side effects of NSAIDs on cystic renal disease progression.
The mechanisms by which COX derived prostanoids modulate cyst growth in the cystic kidney have not yet been defined. The present study showed increased mPGES1 and PGIS expression in the cystic kidney, consistent with the recent report which showed increased levels of PGE2 and PGI2 in the cystic kidney [32]. These findings support a potential role for PGE2 and/or PGI2 in the pathogenesis of cyst growth. In addition, both PGE2 and PGI2 can increase intracellular cAMP levels through the EP2/EP4 and IP receptors respectively, and cAMP has been suggested to promote cyst growth [33]. The EP4 receptor has also been reported to activate the PI3K/AKT pathway, which may lead to activation of mTOR, inhibition of which has been reported to reduce cyst growth [24]. Whether these pathways are responsible for the cyst growth in ADPKD remains to be explored.
Epithelial-stromal interactions and induction of angiogenesis have emerged as important concepts in the pathogenesis of ADPKD [34]. COX2 has been suggested to promote angiogenesis in intestinal polyps and in colon cancer. Whether COX derived prostanoids enhance cyst expansion through their effect on angiogenesis remains to be explored. High levels of mPGES1 were detected in renal cystic epithelial cells where COX1 is normally expressed [35], thus we speculate that PGE2 synthesis from epithelial cells derived via the COX1-mPGES pathway may also be involved in cyst growth.
In conclusion, the present study shows that COX2 expression is increased in cystic kidneys from human ADPKD and a murine Pkd2WS25/- compared to non-cystic control kidneys. Expression of the downstream prostanoids mPGES and PGIS were also notably increased in Pkd2WS25/- kidneys, the significance of which is unclear. COX inhibition with Sulindac significantly reduced kidney size of the Pkd2 mice, suggesting that the COX pathway is a potential therapeutic target for ADPKD.
Supplementary Material
Supplementary figure.
Acknowledgements
Pkd2+/WS25 and Pkd2WS25/WS25 animal models were kindly provided by Drs. Stefan Somlo and Guanqing Wu.
Funding
This study was supported by the National Natural Science Foundation of China (grants 81130075, 31471101 and 81400711) and 985 project 985 III-YFX0302. This study was supported by NIDDK grants DK071876 and DK074116 (to C.M.Hao).
Ethical approval
All animal experiments were performed according to animal protocols approved by the Animal Care Use Committee at Vanderbilt University School of Medicine.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Collins AJ, Foley RN, Herzog C. et al. US Renal Data System 2010 Annual Data Report. Am J Kidney Dis. 2011;57(1 Suppl 1):A8 e1-526
2. Lentine KL, Xiao H, Machnicki G. et al. Renal function and healthcare costs in patients with polycystic kidney disease. Clin J Am Soc Nephrol. 2010;5(8):1471-9
3. Guay-Woodford LM. Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol. 2003;285(6):F1034-49
4. Rossetti S, Consugar MB, Chapman AB. et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18(7):2143-60
5. Grantham JJ. Mechanisms of progression in autosomal dominant polycystic kidney disease. Kidney Int. 1997;63(Suppl):S93-7
6. Rossetti S, Harris PC. Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2007;18(5):1374-80
7. Hao CM, Breyer MD. Roles of lipid mediators in kidney injury. Semin Nephrol. 2007;27(3):338-51
8. Dubois RN, Abramson SB, Crofford L. et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12(12):1063-73
9. Hla T, Bishop-Bailey D, Liu CH. et al. Cyclooxygenase-1 and -2 isoenzymes. Int J Biochem Cell Biol. 1999;31(5):551-7
10. Rostom A, Dube C, Lewin G. et al. Nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force. Ann Intern Med. 2007;146(5):376-89
11. Bernstein J, Evan AP, Gardner KD Jr. Epithelial hyperplasia in human polycystic kidney diseases. Its role in pathogenesis and risk of neoplasia. Am J Pathol. 1987;129(1):92-101
12. Ibrahim NH, Gregoire M, Devassy JG. et al. Cyclooxygenase product inhibition with acetylsalicylic acid slows disease progression in the Han:SPRD-Cy rat model of polycystic kidney disease. Prostaglandins Other Lipid Mediat. 2015;116-117:19-25
13. Nauta J, Goedbloed MA, Luider TM. et al. The Han:SPRD rat is not a genetic model of human autosomal dominant polycystic kidney disease type 1. Lab Anim. 1997;31(3):241-7
14. Wu G, D'Agati V, Cai Y. et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93(2):177-88
15. Lal G, Ash C, Hay K. et al. Suppression of intestinal polyps in Msh2-deficient and non-Msh2-deficient multiple intestinal neoplasia mice by a specific cyclooxygenase-2 inhibitor and by a dual cyclooxygenase-1/2 inhibitor. Cancer Res. 2001;61(16):6131-6
16. Oshima M, Dinchuk JE, Kargman SL. et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell. 1996;87(5):803-9
17. Brideau C, Kargman S, Liu S. et al. A human whole blood assay for clinical evaluation of biochemical efficacy of cyclooxygenase inhibitors. Inflamm Res. 1996;45(2):68-74
18. Breyer MD, Jacobson HR, Davis LS. et al. In situ hybridization and localization of mRNA for the rabbit prostaglandin EP3 receptor. Kidney Int. 1993;44(6):1372-8
19. Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol. 2006;1(1):148-57
20. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287-1301
21. Nauli SM, Alenghat FJ, Luo Y. et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129-37
22. Watnick T, He N, Wang K. et al. Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat Genet. 2000;25(2):143-4
23. Tao Y, Kim J, Schrier RW. et al. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol. 2005;16(1):46-51
24. Wahl PR, Serra AL, Le Hir M. et al. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant. 2006;21(3):598-604
25. Qian F, Watnick TJ, Onuchic LF. et al. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87(6):979-87
26. Roix J, Saha S. TNF-alpha blockade is ineffective in animal models of established polycystic kidney disease. BMC Nephrol. 2013:25 14:233
27. Doctor RB, Serkova NJ, Hasebroock KM. et al. Distinct patterns of kidney and liver cyst growth in pkd2(WS25/-) mice. Nephrol Dial Transplant. 2010;25(11):3496-504
28. Keller JJ, Offerhaus GJ, Hylind LM. et al. Rectal epithelial apoptosis does not predict response to sulindac treatment or polyp development in presymptomatic familial adenomatous polyposis patients. Cancer Epidemiol Biomarkers Prev. 2002;11(7):670-1
29. Giardiello FM, Yang VW, Hylind LM. et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med. 2002;346(14):1054-9
30. Samadder NJ, Neklason DW, Boucher KM. et al. Effect of Sulindac and Erlotinib vs Placebo on Duodenal Neoplasia in Familial Adenomatous Polyposis: A Randomized Clinical Trial. JAMA. 2006;315(12):1266-75
31. Komhoff M, Wang JL, Cheng HF. et al. Cyclooxygenase-2-selective inhibitors impair glomerulogenesis and renal cortical development. Kidney Int. 2000;57(2):414-22
32. Warford-Woolgar L, Peng CY, Shuhyta J. et al. Selectivity of cyclooxygenase isoform activity and prostanoid production in normal and diseased Han:SPRD-cy rat kidneys. Am J Physiol Renal Physiol. 2006;290(4):F897-904
33. Torres VE, Wang X, Qian Q. et al. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med. 2004;10(4):363-4
34. Qian Q, Harris PC, Torres VE. Treatment prospects for autosomal-dominant polycystic kidney disease. Kidney Int. 2001;59(6):2005-2022
35. Qi Z, Hao CM, Langenbach RI. et al. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J Clin Invest. 2002;110(1):61-9
Author contact
![]() Corresponding author: Chuan-Ming Hao, Division of Nephrology, Huashan Hospital, Fudan University, Shanghai, China. Email: chuanminghaoedu.cn
Corresponding author: Chuan-Ming Hao, Division of Nephrology, Huashan Hospital, Fudan University, Shanghai, China. Email: chuanminghaoedu.cn