Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(1):167-179. doi:10.7150/ijms.24068 This issue Cite
Research Paper
Lipopolysaccharide promoted proliferation and adipogenesis of preadipocytes through JAK/STAT and AMPK-regulated cPLA2 expression
Chao-Chien Chang1,2,3,4, Kee-Chin Sia5, Jia-Feng Chang5,6,7, Chia-Mo Lin5,8,9, Chuen-Mao Yang10,11,12, Kuo-Yang Huang13, Wei-Ning Lin5 ![]()
1. Division of Cardiology, Department of Internal Medicine, Cathay General Hospital, Taipei, Taiwan;
2. Graduate Institute of Medical Sciences, College of Medicine, Taipei Medical University, Taipei, Taiwan;
3. Department of Pharmacology, School of medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan;
4. School of Medicine, College of Medicine, Fu Jen Catholic University, New Taipei City, Taiwan;
5. Graduate Institute of Biomedical and Pharmaceutical Science, Fu Jen Catholic University, New Taipei City, Taiwan;
6. PhD Program in Nutrition and Food Science, Fu Jen Catholic University, New Taipei City, Taiwan;
7. Department of Internal Medicine, En-Chu-Kong Hospital, New Taipei City, Taiwan;
8. Department of Chemistry, Fu-Jen Catholic University, New Taipei, Taiwan;
9. Division of Chest Medicine, Shin Kong Hospital, Taipei, Taiwan;
10. Department of Physiology and Pharmacology and Health Ageing Research Center, College of Medicine, Chang Gung University, Kwei-San, Tao-Yuan, Taiwan;
11. Department of Anesthetics, Chang Gung Memorial Hospital at Linkuo and Chang Gung University, Kwei-San, Tao-Yuan, Taiwan:
12. Research Center for Chinese Herbal Medicine and Research Center for Food and Cosmetic Safety, College of Human Ecology, Chang Gung University of Science and Technology, Tao-Yuan, Taiwan;
13. Graduate Institute of Pathology and Parasitology, National Defense Medical Center, Taipei, Taiwan
Received 2017-11-28; Accepted 2018-12-4; Published 2019-1-1
Abstract
The proliferation and adipogenesis of preadipocytes played important roles in the development of adipose tissue and contributed much to the processes of obesity. On the other hand, lipopolysaccharide (LPS), also known as endotoxin, is a key outer membrane component of gram-negative bacteria in the gut microbiota, and has a dominant role in linking inflammation to high-fat diet-induced metabolic syndrome. Studies suggested the potential roles of LPS in hepatic steatosis and in obese mice models. However, the molecular mechanisms underlying LPS-regulated obesity remained largely unknown. Here we reported that LPS stimulated expression of cyosolic phospholipase A2 (cPLA2), one of inflammation regulators of obesity, in the preadipocytes. Pretreatment the inhibitors of JAK2, STAT3, STAT5 or AMPK significantly reduced LPS-increased mRNA and protein expression of cPLA2 together with phosphorylation of JAK2, STAT3, STAT5 and AMPK, separately. Similarly, transfection of siRNA against JAK2 or AMPK abolished expression of cPLA2 and phosphorylation of JAK2 or AMPK together with downregulated expression of JAK2 and AMPK protein. LPS enhanced activation of STAT3 and STAT5 via JAK2-dependent manner in the preadipocytes. Transfection of JAK2 or AMPK siRNA further proofed the independence of JAK2 and AMPK in LPS-treated preadipocytes. In addition, LPS-increased DNA synthesis, cell numbers and cell viability of preadipocytes were attenuated by AACOCF3, AG490, BML-275, cPLA2 siRNA, JAK2 siRNA or AMPK siRNA. Attenuation JAK2/STAT or AMPK-dependent cPLA2 expression reduced LPS-mediated adipogenesis of preadipocytes. Stimulation of arachidonic acid or AMPK activator, A-769662, increased cell numbers and cell viability and promoted differentiation of preadipocytes. Collectively, these results indicated that LPS increased preadipocytes proliferation and adipogenesis via JAK/STAT and AMPK-dependent cPLA2 expression. The mechanisms of LPS-stimulated cPLA2 expression may be a link between bacteria and obesity and provides the molecular basis for preventing metabolic syndrome or hyperplasic obesity.
Keywords: cPLA2, Lipopolysaccharide, Adipocyte, Proliteration, Adipogenesis
Introduction
Obesity, defined as “abnormal or excessive fat accumulation, is a chronic disease and a worldwide epidemic problem. Obesity contributes to the development of a group of potentially life-threatening conditions including, insulin resistance, Type 2 Diabetes Mellitus, dyslipidemia, cardiovascular disease, metabolic syndrome, nonalcoholic fatty liver disease, osteoarthritis, stillbirth, and some cancer [1-3]. Adipose tissue consists of approximately one-third of mature adipocytes and two-thirds of stromal cells including macrophages, fibroblasts, endothelial cells and preadipocytes [4]. Preadipocytes originate from a multi-potent stem cell of mesodermal origin and function as source of new fat cells persists during the entire human life. The cellular changes of adipose tissue in obesity include fat depot hypertrophy (increase in adipocyte volumes) and hyperplasia (increase in adipocyte numbers) [5, 6]. Excess triglyceride accumulation in existing adipocytes due to a positive energy balance (energy intake in excess of energy expenditure) results in hypertrophy. On the other way, hyperplasia, regarded as 'adipogenesis', results from the recruitment of new adipocytes from precursor cells in adipose tissue and involves the proliferation and differentiation of preadipocytes [5]. Because increase in adipocyte number from preadipocyte proliferation and differentiation may result in more units to storage lipid. Hyperplastic fat expansion with poorest prognosis for treatment is addressed as more important than hypertrophic expansion [5].
The proliferation of preadipocytes was tightly regulated. In addition to the action of various hormone, several cytokines such as transforming growth factor-β (TGFβ), tumor necrosis factor-α (TNF-α), macrophage colony-stimulating factor (MCSF), angiotensin II, basic fibroblast growth factor (bFGF) and bone morphogenetic protein (BMP) are reported to positively or negatively regulating adipocyte proliferation [7-16]. Lipopolysaccharide (LPS), also known as endotoxin, is a key component of the outer membranes of gram-negative bacteria and has a dominant role in the host responding to gram-negative bacterial infection. It is proposed that LPS derived from gram-negative bacteria residing in the gut microbiota acts as a triggering factor linking inflammation to high-fat diet-induced metabolic syndrome [17]. Studies found that a high-fat diet in mice increases endotoxemia and affect intestinal bacterial populations by favoring an increase in the gram-negative to gram-positive ratio. And chronic metabolic endotoxemia induces obesity, insulin resistance, and diabetes [17]. Similarly, treatment of rats with polymyxin B, an antibiotic that specifically targets gram-negative organisms, is shown to reduce LPS concentration and hepatic steatosis [18]. In culture system, LPS stimulates the expression and secretion of serum amyloid A, LPS binding protein, soluble CD4 and RANTES (a chemokine) in adipocytes [19]. Similar results also show in ob/ob mice or high-fat-diet mice model that intravenous injection of LPS increase the level of serum amyloid A, LPS binding protein, soluble CD4 and RANTES in plasma [19]. Although several reports implied the participation of LPS on obesity, the cellular mechanisms are still largely unknown. Moreover, the role of AMP-activated protein kinase (AMPK) in preadipocytes proliferation and adipogenesis is controversial. It is found that AMPK inhibitor cannot prevent the inhibition effects of EGCG on insulin growth factor-stimulated preadipocyte proliferation [20]. However, AMPK siRNA reversed ursolic acid-inhibited adipogenesis [21]. Thus, AMPK differently contributed to the preadipocytes proliferation and adipogenesis. Whether AMPK involved in LPS-regulated preadipocytes proliferation and adipogenesis was less evaluated.
Cytosolic phospholipase A2 (cPLA2), one of inflammation regulators, contributes to inflammation via upregulating the production of arachidonic acid (AA) and the following eicosanoid. It is found that expression of cPLA2 facilitates the infiltration of neutrophils into adipose tissue [22]. cPLA2 contributes to the process of adipogenesis by promoting the proliferation of preadipocytes and cell cycle progress [23]. Expression of cPLA2 is regulated by Janus tyrosine kinase (JAK)2 that inhibition of JAK2 activation by AG490 abolishes TNF-α and IL-5-regulated cPLA2 expression in human pulmonary alveolar epithelial cells and eosinophils, separately [24, 25]. Also, AG490 suppresses the phosphorylation of signal transducer and activator of transcription (STAT)-3, and both AG490 and dominant-negative mutant of STAT-3 attenuated expression of cPLA2, AA release, and DNA synthesis in PDGF-BB-stimulated vascular smooth muscle cells [26]. In addition, STAT-5 is also reported to be one of JAK2 downstream molecules that may opsonize the effects of LPS [27]. It is found that NVP-BSK805, a specific JAK2 inhibitor, suppressed STAT5 phosphorylation and microglia survival in response to LPS [28]. Whether activation of JAK/STAT pathway involved in LPS-regulated preadipocytes proliferation and adipogenesis was less studied.
In this study, we determined the effects of LPS on preadipocytes proliferation and adipogenesis together with the related molecular mechanisms. Here we reported that LPS increased expression of cPLA2 gene via activation of AMPK and JAK/STAT pathway. Suppression the phosphorylation of AMPK and JAK2 or inhibition of cPLA2 attenuated LPS-stimulated preadipocytes proliferation and adipogenesis. Collectively, LPS contributed to hyperplasic obesity via AMPK and JAK/STAT-dependent activation of cPLA2 gene.
Materials and methods
Materials
Fetal bovine serum (FBS), DMEM medium, and TRIZOL were purchased from Invitrogen (Carlsbad, CA, USA). Antibodies against cPLA2 (SC-454), p-JAK2 (SC-21870), p-STAT3 (SC-8059), p-STAT5 (SC-101806) and GAPDH (SC-32233) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). PhosphoPlus AMPK antibody (#43705) kits were obtained from New England Biolabs (Beverly, MA, USA). AG490, WP1066, STAT5-I, BML-275 and AACOCF3 were obtained from Biomol (Plymouth Meeting, PA, USA). Hybond C membrane and Hyperfilms were obtained from GE Healthcare Biosciences (Buckinghamshire, UK). siRNA of scrambled, AMPK, JAK2, and cPLA2 were purchased from MDBio, Inc. (Taipei, Taiwan). An enhanced chemiluminescence (ECL) Western blotting detection system was obtained from Visual Protein Biotechnology Co. (Taipei, Taiwan). XTT assay kit was purchased from Biological Industries (Beth-Haemek, Israel). LPS, enzymes and other chemicals were obtained from Sigma (St. Louis, MO, USA).
Cell culture and adipogenesis
3T3-L1 preadipocytes were purchased from Food Industry Research and Development Institute (Hsinchu, Taiwan) and cultured in 37 °C, 5% CO2 with DMEM medium containing 10% FBS. Differentiation of preadipocytes to adipocytes was induced by incubating cells in differentiation medium (DM)-I (DMEM medium containing 0.5 mM of methylisobutylxanthine, 1 μg/ml of insulin, 0.25 μM of dexamethasome) for 48 h. And then cells were changed to DM-II medium (DMEM medium containing 1 μg/ml of insulin) for another 2 to 6 days. Mature adipocytes was confirmed by Oil Red O (from sigma)-stained fat droplets in the cytoplasm.
Oil Red O stain
At the end of differentiation, adipocytes were washed with PBS and fixed with 10% formalin by incubating 1 h at RT. At the end of incubation, formalin was removed and washed with 60% isopropanol once. Then Oil Red O working solution (from invitrogen) was added into cells for 10 min then washed out by H2O four times. Lipids appeared in red and cells were viewed on a phase contrast microscope (DMI 3000 B; Leica, Wetzlar, Germany). For quantification the amount of lipid in adipocytes, Oil Red O was eluted by adding 100% isopropanol for 10 min. After pipet up and down to sure that all Oil Red O was in the solution, the isopropanol with Oil Red O was transfer to 96-well plate and measure OD at 490 nm by Epoch™ Multi-Volume Spectrophotometer System (BioTek, Vermont, USA). 100% isopropanol was used as blank control.
Transfection with small interference RNA (siRNA)
3T3-L1 cells were plated in 3 X 105 cells/mL (1 mL/well) in 12-well culture plates for 24 h, reaching approximately 80% confluence [29]. The cells were replaced with 0.4 mL of DMEM containing 10% FBS. The DNA Metafectene reagent complex was prepared according to manufacturer instructions (Biontex, Martinsried, Planegg, Germany). The amount of transfected DNA was maintained constant with 100 nM scrambled, AMPK, JAK2, or cPLA2 siRNA for each well. The sense sequences of siRNA used are as follows: JAK2: CGGGUCGGCGCAACCUAAGAUUAAU; AMPKα: AUGAUGUCAGAUGGUGAAUUU; cPLA2: CGAGACACUUCAAUAAUGAUU; and scramble: UUCUCCGAACGUGUCACGU. The DNA METAFECTENE complex (0.1 mL) was added to each well and then incubated at 37 °C for 24 h. After 24 h of transfection, the cells were washed with PBS and maintained in DMEM medium for 72 h (before treatment with LPS for the indicated time intervals).
Cell lysate extraction and Western blot
After treatment, the cells were then rapidly washed with ice-cold PBS, scraped, and collected by centrifugation at 1,000 g for 10 min. The collected cells were lysed with ice-cold lysis buffer. The lysates were centrifuged at 4,500 g for 1 h at 4 °C to yield the whole cell extract. Samples from these supernatant fractions (30 μg protein) were subjected to SDS-PAGE using a 10% running gel. Proteins were transferred to nitrocellulose membrane, and the membrane was incubated successively at room temperature with 5% BSA in Tris-buffered saline with 0.1% Tween 20 (TTBS) for 1 h. Membranes were incubated overnight at 4°C with an anti-cPLA2, anti-COX-2, anti-phospho-AMPK, anti-phospho-JAK2, anti-phospho-STAT3, anti-phospho-STAT5, or anti-GAPDH according to the recommendation of the manufacturer. Membranes were incubated with a 1:2,000 dilution of anti-mouse or anti-rabbit horseradish peroxidase antibody for 1 h. The immunoreactive bands detected by ECL reagents were developed by Hyperfilm-ECL.
RNA extraction and semi-quantified PCR
Total RNA was extracted from 3T3-L1 cells using Trizol, as previously described [30]. The cDNA containing 2 μg of RNA was used as a template to analyze cPLA2 mRNA level. Oligonucleotide primers for β-actin and cPLA2 were as follows: for β-actin: 5'-GGCAT TGTTA CCAAC TGGGA CGAC-3' (sense), 5'-GGCAT TGTTA CCAAC TGGGA CGAC-3' (antisense); for cPLA2: 5'-GTGAG GGGCT TTATT CCACA-3' (sense), 5'-GGTGA GAGTA CAAGG TTGAC A-3' (antisense). The amplification profile included one cycle of initial denaturation at 94 °C for 5 min, 30 cycles of denaturation at 94 °C for 1 min, primer annealing at 55 °C (cPLA2) and 60 °C (β-actin) for 1 min, extension at 72 °C for 1 min, and then one cycle of final extension at 72 °C for 5 min. The expression of β-actin was used as an internal control for the assay of a constitutively expressed gene.
Cell viability assay and cell number counts
The working solution of XTT assay kit was prepared as manufacturer's direction. The Cultured cells (5000 cells/well) were treated with or without various inhibitors and then incubated with LPS for 48 h. At the end of incubation, 50 μL of XTT kit reaction solution was added into each well and incubated in an incubator for 2 h. The absorbance of each well was detected at OD450 and OD630 (reference absorbance) by Epoch™ Multi-Volume Spectrophotometer System (BioTek, Vermont, USA). Or cells were cultured in 6-cm dishes, and incubated with various treatments. At the end of stimulation, cell numbers were counted by HoloMonitor M4 (Phase Holographic Imaging PHI AB, Lund, Sweden).
Bromodeoxyuridine (BrdU) incorporation assay
10 μM BrdU labeled cells were pretreated with various inhibitors and then incubated with 20 μg/mL of LPS for 48 h. At the end of treatment, cells were washed 3 times with PBS followed by methanol fixation and permeabilization for 20 minutes at -20°C. After washing cells, anti-BrdU antibody (1:100) was added for 1 h at 37°C. After washes, the FITC-conjugated secondary antibody were added at 1:200 at 37°C for 1 h. Cells were visualized under a fluorescence microscope (DMI 3000 B; Leica, Wetzlar, Germany).
Statistical Analysis of Data
All data are expressed as the mean ± standard error of the mean by using the GraphPad Prism Program (GraphPad, San Diego, CA, USA) [30]. Quantitative data were analyzed using one-way ANOVA followed by Tukey's post hoc test at a p < 0.05 level of significance. All of the experiments were performed at least 5 times.
Results
LPS stimulated expression of cPLA2 in preadipocytes
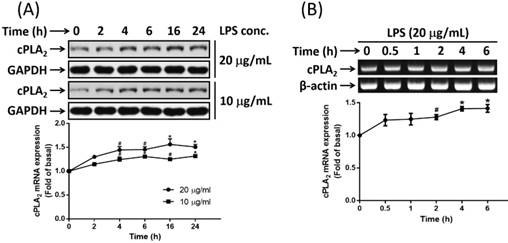
It is reported that endotoxemia occurs in high-fat diet-fed mice and chronic metabolic endotoxemia induces obesity, insulin resistance, and diabetes [17]. LPS, also known as endotoxin, is the key component of the outer membranes of gram-negative bacteria and plays important roles in inducing host responses against infection. On the other hand, cPLA2 shows a proadipogenic function in regulating the process of adipogenesis [23]. The ability whether LPS promoted cPLA2 expression in preadipocytes were determined. Low serum-growth arrested cells were stimulated by 20 or 10 μg/mL of LPS for 0, 2, 4, 6, 16 or 24 h. At the end of incubation, cells were washed and cell lysates were extracted. Protein lysates were subjected into 10% SDS-PAGE and Western blot was performed with the usage of anti-cPLA2 antibody. We found that LPS stimulated expression of cPLA2 protein in a time-dependent manner with maximum response occurred after 16 h of stimulation (Fig. 1A). In addition, to detect whether LPS mediated cPLA2 mRNA expression in preadipocytes, cells were treated with 20 μg/mL of LPS for 0, 0.5, 1, 2, 4 or 6 h. RT-PCR was performed to detect the mRNA expression of cPLA2. As showed in Fig. 1B, LPS induced increased expression of cPLA2 mRNA in preadipocytes with maximum responses at the end of studied time point. Thus, LPS increased cPLA2 gene expression in preadipocytes.
LPS enhanced cPLA2 gene expression in 3T3-L1 cells. Serum-starved 3T3-L1 cells were stimulated with different concentrations of LPS for the indicated time points. At the end of incubation, cells were harvested and cell lysates or mRNA were extracted. (A) Western blot was used to evaluate the expression of cPLA2 protein. (B) RT-PCR was used to analyze the expression of cPLA2 mRNA. Data are expressed as means ± SEM of at least 3 independent experiments (n≥3). #P < 0.01, *P < 0.05, as compared with the basal group.
LPS mediated cPLA2 expression via activation of JAK2 kinase
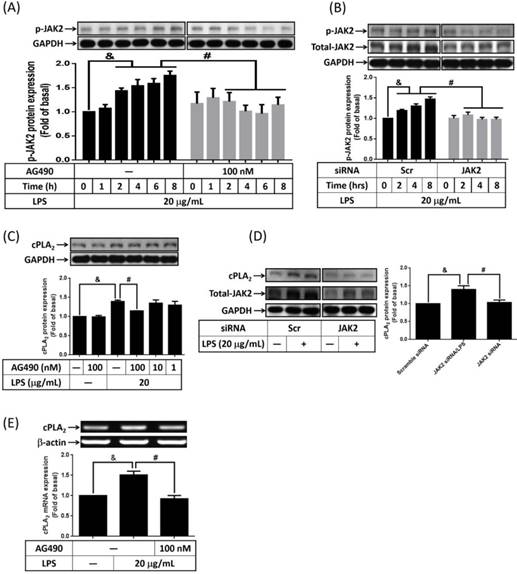
It is reported that expression of cPLA2 gene is mediated by TNF-α or IL-5-increased JAK2 activity [24, 25]. To elucidate whether LPS increased phosphorylation of JAK2 in 3T3-L1 cells, serum-starved cells were treated with 20 μg/mL of LPS for 0, 1, 2, 4, 6 or 8 h. Phosphorylation of JAK2 was detected by Western blot with anti-phospho-JAK2 antibody. Phosphorylation of JAK2 began as early as 2 h after LPS stimulation, and sustained to 8 h. Pretreatment of AG490, inhibitor of JAK2, significantly attenuated LPS-regulated JAK2 phosphorylation in 3T3-L1 cells (Fig. 2A). Similarly, transfection of JAK2 siRNA down-regulated JAK2 protein expression together with abolished JAK2 phosphorylation in LPS-stimulated preadipocytes (Fig. 2B). To evaluate whether JAK2 involve in LPS-stimulated cPLA2 expression, cells were pretreated with AG490 for 1 h. After treated with 20 μg/mL of LPS for another 16 h, cell lysates were collected and subjected into 10% SDS-PAGE. As showed in Fig. 2C, AG490 significantly attenuated LPS-induced cPLA2 expression. Transfection of JAK2 siRNA also reduced LPS-regulated cPLA2 expression (Fig. 2D). Similarly, blockage JAK2 by AG490 obviously reduced LPS-regulated cPLA2 mRNA expression (Fig. 2E). These data suggested that LPS enhanced cPLA2 gene expression via activation of JAK2 in preadipocytes.
LPS modulated cPLA2 gene expression via activation of JAK2. Serum-starved 3T3-L1 cells were pretreated with 100 nM or different concentrations of AG490 for 1 h. Or cells were transfected with 100 nM of scramble (Scr) or JAK2 siRNA for 24 h. Inhibitor or siRNA-treated cells were then incubated 20 μg/mL of LPS for the indicated time points (A, B), 16 h (C, D) or 6 h (E). At the end of incubation, cells were harvested and cell lysates or mRNA were extracted. (A, B, C, D) Western blot was used to evaluate the expression of phosphorylated JAK2, total JAK2, cPLA2 or GAPDH protein. (E) RT-PCR was used to analyze the expression of cPLA2 mRNA. Data are expressed as means ± SEM of at least 3 independent experiments (n≥3). &P < 0.05, as compared with the 0 point or indicated group. #P < 0.05, as compared with the same time points or LPS treated alone.
Involvement of STAT3 in LPS-stimulated cPLA2 expression
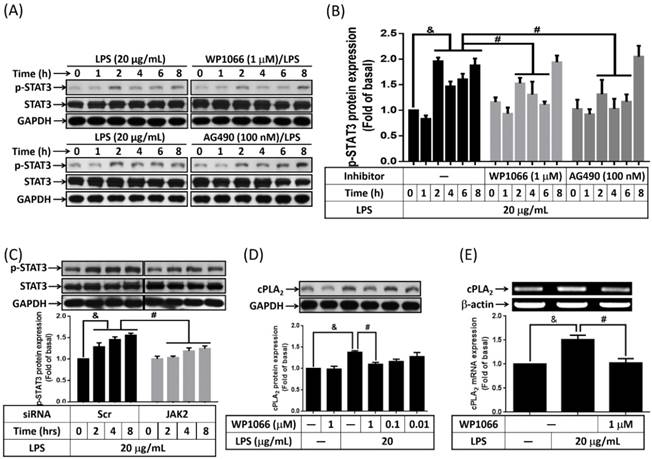
STAT3 is one of JAK2 downstream signaling molecular that regulating PDGF-BB-stimulated cPLA2 expression in vascular smooth muscle cells [26]. Whether LPS increased cPLA2 expression via JAK2-dependent activation of STAT3 was investigated in preadipocytes. Serum-starved 3T3-L1 cells were stimulated by 20 μg/mL of LPS for 0, 1, 2, 4, 6, or 8 h. The phosphorylation of STAT3 was detected by Western blot with anti-phospho-STAT3 antibody. LPS increased STAT phosphorylation was detected as early as 2 h after stimulation. Pretreatment of WP1066 (inhibitor of STAT3) or AG490 significantly decreased STAT3 phosphorylation in LPS-stimulated cells (Fig. 3A and B). Moreover, JAK2 siRNA transfection decreased STAT3 phosphorylation level in LPS-treated preadipocytes (Fig. 3C). To ensure the role of STAT3 in regulating LPS-induced cPLA2 gene expression, cells were pretreated with different concentrations or 1 μM of WP1066 for 1 h, then incubated with LPS for 16 h or 6h. We found that LPS-stimulated cPLA2 protein expression was significantly reduced by WP1066 (Fig. 3D). Similarly, inhibition of STAT3 by WP1066 significantly attenuated cPLA2 mRNA expression in LPS-treated cells (Fig. 3E). Briefly, these data suggested that the participation of STAT3 in LPS-stimulated cPLA2 expression.
Activation of STAT3 contributed to LPS-regulated cPLA2 gene expression. Serum-starved 3T3-L1 cells were pretreated with AG490 (100 nM) or WP1066 (1 μM or different concentration) for 1 h. Or cells were transfected with 100 nM of scramble (Scr) or siRNA for 24 h. At the end of inhibitor or siRNA treatment, cells were incubated 20 μg/mL of LPS for the indicated time points (A, B, C), 16 h (D) or 6 h (E). Cells were harvested and cell lysates or mRNA were extracted. (A, C, D) Western blot was used to evaluate the phosphorylation of STAT3, total STAT3 or cPLA2 protein. (B) The phosphorylated STAT3 was quantified and showed as bar graph. (E) RT-PCR was used to analyze the expression of cPLA2 mRNA. Data are expressed as means ± SEM of at least 3 independent experiments (n≥3). &P < 0.05, as compared with the 0 point group or the indicated group. #P < 0.05, as compared with the same time points or LPS treated alone.
Activation of STAT5 in LPS-increased cPLA2 expression
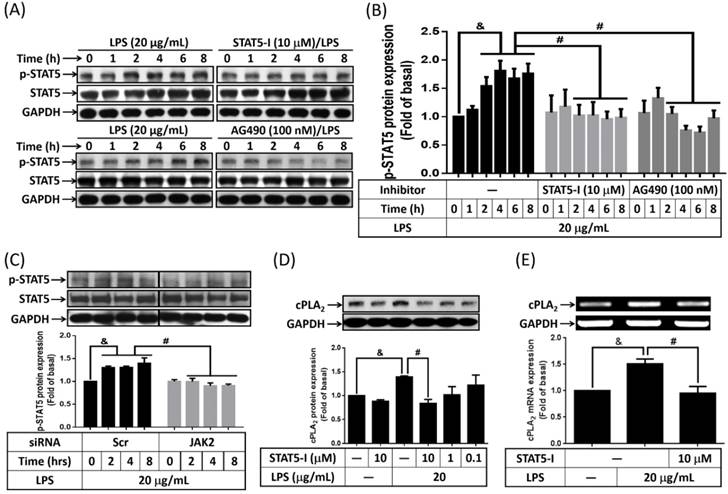
Activation inhibition of STAT5 by peroxiredoxin V-dependent suppression of JAK2 attenuated LPS-induced immune response [27]. Whether LPS activated STAT5 in preadipocytes was studied. Cells were treated with or without AG490 or STAT5-I (inhibitor of STAT5) for 1h, and then stimulated by 20 μg/mL of LPS for the indicated time intervals. Phosphorylation of STAT5 was detected by Western blot. As showed in Fig. 4A and B, LPS increased phosphorylation of STAT5 as early as 2 h after treatment, and sustained to 8 h. Pretreatment of STAT5-I and AG490 significantly attenuate LPS-induced phosphorylation of STAT5 in preadipocytes (Fig. 4A and B). LPS-increased phosphorylation of STAT5 also be reduced by transfection of JAK2 siRNA (Fig. 4C). To know whether activated STAT5 contributed to LPS-increased expression of cPLA2 gene, cells were pretreated with various concentrations or 10 μM of STAT5-I for 1 h, and incubated with LPS for 16 or 6 h. Protein lysates or mRNA were extracted and analyzed by Western blot and RT-PCR, separately. LPS-enhanced expression of cPLA2 protein and mRNA was significantly reversed by STAT5-I (Fig. 4D and E). Collectively, these data revealed that LPS regulated cPLA2 gene expression via activation of JAK/STAT5 pathway.
Participation of STAT5 in LPS-mediated cPLA2 gene expression. Serum-starved 3T3-L1 cells were pretreated with AG490 (100 nM) or STAT5-I (10 μM or different concentration) for 1 h. Or cells were transfected with 100 nM of scramble (Scr) or JAK2 siRNA for 24 h. After inhibitor or siRNA treatment, cells were incubated with 20 μg/mL of LPS for the indicated time points (A, B, C), 16 h (D) or 6 h (E). Protein lysates or mRNA were extracted. (A, C, D) Western blot was used to evaluate the phosphorylation of STAT5, total STAT5 or cPLA2 protein. (B) The phosphorylated STAT5 was quantified and showed as bar graph. (E) RT-PCR was used to analyze the expression of cPLA2 mRNA. Data are expressed as means ± SEM of at least 3 independent experiments (n≥3). &P < 0.05, as compared with the 0 point group or the indicated group. #P < 0.05, as compared with the same time points or LPS treated alone.
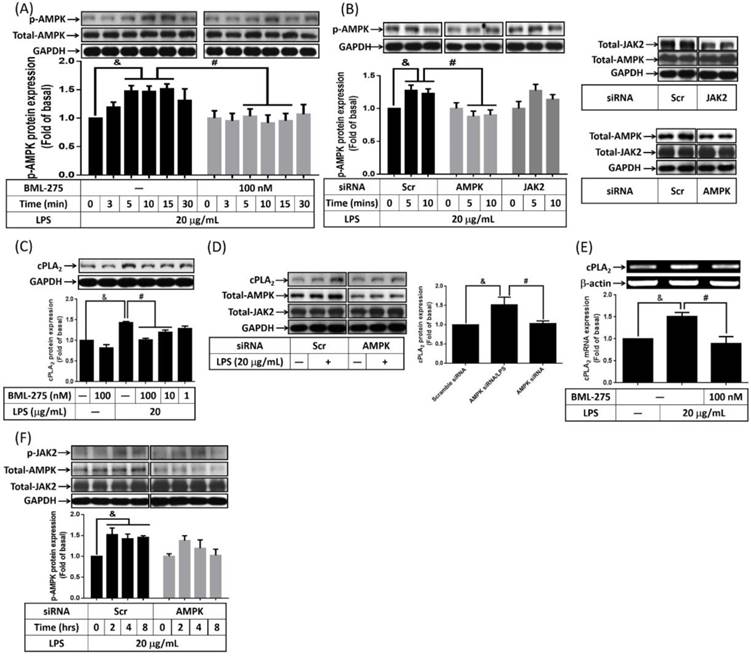
Participation of AMPK in LPS-enhanced cPLA2 expression
The role of AMPK in LPS-promoted hyperplasia obesity is controversial. It is reported that inhibition of AMPK reversed ursolic acid but not EGCG effects of adipogenesis [20, 21]. The effects of LPS on AMPK activity was examined on preadipocytes, cells were pretreated with AMPK inhibitor, BML-275, for 1 h, and then incubated with LPS for the indicated time points. Or AMPK siRNA transfected cells were incubated with 20 μg/mL of LPS for the indicated time points. After harvested, cells lysates were subjected into 10% SDS-PAGE. Western blot was performed with the usage of anti-phospho-AMPK. LPS increased phosphorylation of AMPK in preadipocytes, which was significantly attenuated by BML-275 (Fig. 5A). Similarly, LPS-increased AMPK phosphorylation was abolished by knockdown AMPK (Fig. 5B). To delink the relationship between AMPK and cPLA2 expression in LPS-regulated preadipocytes, cells were pretreated with different concentrations or 100 nM of BML-275 for 1 h or cells were transfected with 100 nM scramble or AMPK siRNA. After treated with 20 μg/mL of LPS, cell lysates or mRNA were collected and analyzed. LPS-increased expression of cPLA2 was significantly attenuated by BML-275 (Fig. 5C) or AMPK siRNA (Fig. 5D). Similarly, pretreatment of BML-275 reversed cPLA2 mRNA expression in LPS-stimulated preadipocytes (Fig. 5E). These data suggested that LPS induced cPLA2 gene expression via activation of AMPK in preadipocytes. To distinguish the relationship between AMPK and JAK2 activation in LPS-treated cells. Preadipocytes were transfected with scramble, AMPK or JAK2 siRNA and then incubated with LPS for the indicated time points. Knockdown AMPK protein did not attenuated LPS-increased JAK2 phosphorylation (Fig. 5F). In addition, LPS-stimulated AMPK phosphorylation did not affected by knock down JAK2 protein (Fig. 5B), suggested the independence of AMPK and JAK2 in LPS-treated preadipocytes. Collectively, LPS enhanced cPLA2 gene expression via two manners, JAK2 and AMPK.
LPS increased cPLA2 gene expression through activation of AMPK. Serum-starved 3T3-L1 cells were pretreated with BML-275 (100 nM or different concentration) for 1 h or transfected with 100 nM of scramble (Scr) or AMPK siRNA for 24 h, then incubated 20 μg/mL of LPS for the indicated time points (A, B, F), 16 h (C, D) or 6 h (E). At the end of incubation, cells were harvested and cell lysates or mRNA were extracted. (A, B, C, D, F) Western blot was used to evaluate the expression of p-AMPK, total-AMPK, p-JAK2, total-JAK2, cPLA2 or GAPDH. (E) RT-PCR was used to analyze the expression of cPLA2 mRNA. Data are expressed as means ± SEM of at least 3 independent experiments (n≥3). &P < 0.05, as compared with the 0 point group or control group. #P < 0.05, as compared with the same time points or LPS treated alone.
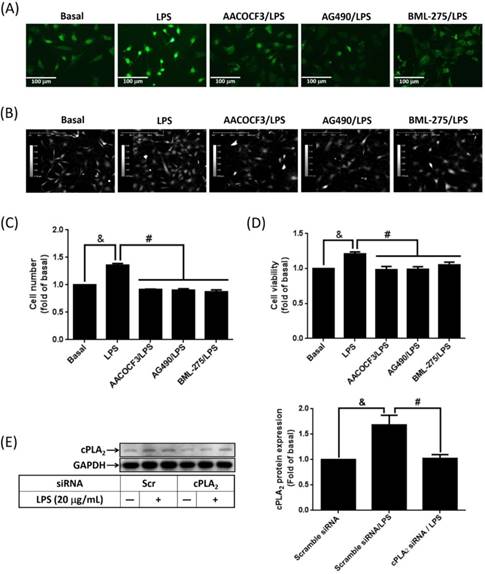
Attenuation of LPS-increased cell proliferation by blockage JAK2/AMPK-dependent cPLA2 expression
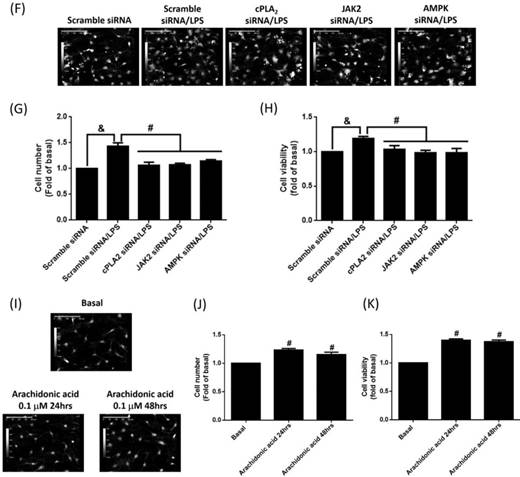
To study whether LPS-increased cPLA2 gene expression contributed to proliferation of preadipocytes, 10 μM BrdU-labeled cells were pretreated with AACOCF3, inhibitor of cPLA2 for 1 h, and then stimulated by LPS for 48 h. The BrdU incorporation was observed by fluorescence microscope. LPS increased the DNA synthesis or preadipocytes, which was reversed by AACOCF3 (Fig. 6A). This suggested that LPS facilitated preadipocyte proliferation via cPLA2 protein. Similarly, pretreatment of AG490 and BML-275 both attenuated LPS-stimulated DNA synthesis in preadipocytes (Fig. 6A). In the aspect of cell counts, pretreatment of AACOCF2, AG490 or BML-275 significantly reduced LPS-increased cell numbers of preadipocytes (Fig. 6B and C). Cell viability assay also revealed that LPS increased the viability of preadipocytes, which was significantly reduced by pretreatment of AACOCF3, AG490 or BML-275 (Fig. 6D). To ascertain whether LPS promoted cell proliferation via JAK2 and AMPK-regulated cPLA2 gene, cells were transfected with siRNA of scramble, cPLA2, JAK2 or AMPK, and then incubated with LPS. Transfection of cPLA2 siRNA significantly reduced cPLA2 expression in LPS-treated cells (Fig. 6E). We found that LPS-increased cell numbers and viability were significantly reversed by knockdown expression of cPLA2, JAK2 or AMPK protein (Fig. 6F, G and H). Furthermore, Application of arachidonic acid increased cell numbers and viability of preadipocytes (Fig. 6I, J and K). These data revealed that LPS enhanced proliferation of preadipocytes via JAK2 and AMPK-regulated cPLA2 protein.
LPS enhanced proliferation of preadipocytes via JAK2 and AMPK-regulated cPLA2 protein. Serum-starved 3T3-L1 cells were pretreated with AACOCF3 (1 μM), AG490 (100 nM) or BML-275 (0.1 μM) for 1 h. Or cells were transfected with siRNA against scramble, JAK2, AMPK or cPLA2 for 24 h. Then cells were incubated 20 μg/mL of LPS 48 h. Or, cells were incubated with 0.1 μM of arachidonic acid for 0, 24 or 48 h. At the end of incubation, (A) BrdU incorporation assay, (B, F, I) HoloM4 system detection, (C, G, J) cell number counts, and (D, H, K) XTT assay were performed. (E) Western blot was used to detect the expression of cPLA2. Data are expressed as means ± SEM of at least 3 independent experiments (n≥3). &P < 0.05, as compared with the control group or scramble siRNA transfected alone group. #P < 0.05, as compared with the LPS treated alone or the basal group.
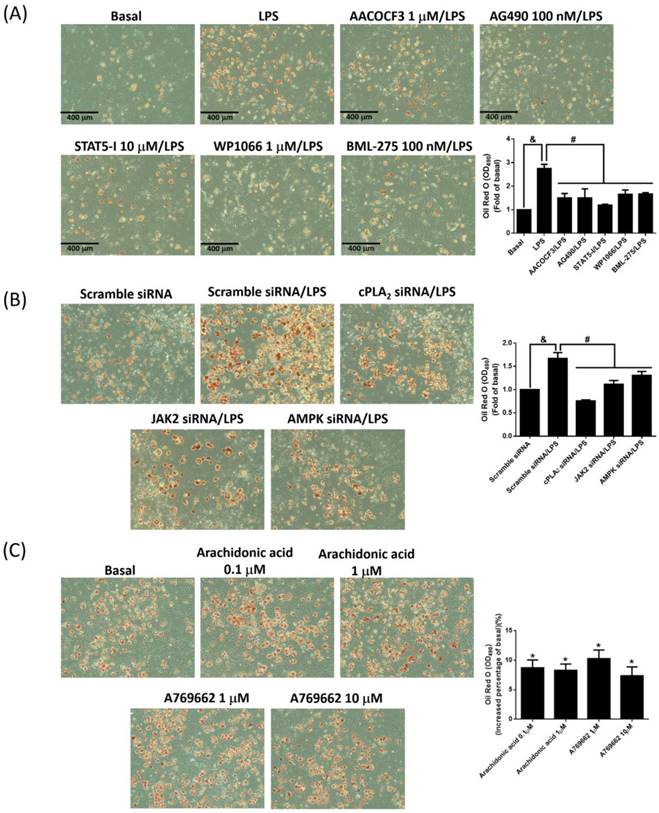
To evaluate whether LPS accelerated adipogenesis via JAK2/STAT and AMPK-dependent cPLA2 expression, adipogenesis assay were performed with the addition of LPS alone or coexistence of various inhibitors or siRNAs. LPS increased the rates of adipogenesis, and attenuation of cPLA2 activity reduced adipogenesis in LPS-stimulated preadipocytes (Fig. 7A). Consistently, inhibition cPLA2 expression by blockage JAK2/STAT or AMPK pathway also reversed LPS-increased adipogenesis (Fig. 7A). On the aspect of knockdown experiments, LPS-promoted adipogenesis were significantly blockage by protein knockdown of cPLA2, JAK2 or AMPK (Fig. 7B). Moreover, application of arachidonic acid or AMPK activator alone slightly increased the adipogenesis of preadipocytes (Fig. 7C). In summary, these data indicated that LPS increased proliferation and adipogenesis of preadipocytes via JAK2/STAT and AMPK-regulated cPLA2 expression.
LPS promoted adipogenesis via JAK/STAT and AMPK-dependent cPLA2 expression. (A) Serum-starved 3T3-L1 cells were pretreated with AACOCF3 (1 μM), AG490 (100 nM), STAT5-I (10 μM), WP1066 (1 μM) or BML-275 (0.1 μM) for 1 h. (B) Or cells were transfected with siRNA of scramble, cPLA2, JAK2 or AMPK fro 24 h. Then, cells were incubated with 20 μg/mL of LPS 48 h. Or (C) cells were stimulated without or with arachidonic acid (0.1 or 1 μM) or A769662 (1 or 10 μM) for 24 h. At the end of incubation, the adipogenesis was performed with DM-I and DM-II medium. After the process of adipogenesis, the images were captured by microscope. Data are expressed as means ± SEM of at least 3 independent experiments (n≥3). &P < 0.05 or *P < 0.05, as compared with the control group or scramble siRNA alone group. #P < 0.05, as compared with the LPS treated alone.
Proposed model underlying LPS regulated preadipocyte proliferation and adipogenesis. LPS increased preadipocyte proliferation and adipogenesis via induction of inflammatory gene, cPLA2, expression. Series studies revealed that LPS stimulated cPLA2 expression via activation JAK/STAT and AMPK in preadipocytes.
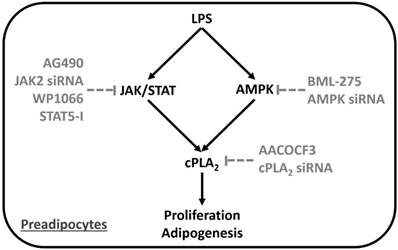
Discussion
The obese problem of the world was getting worse, but the drug against weight gain was lack. The proliferation and adipogenesis of preadipocytes were the cellular changes of hypertrophic obesity. It is found that inflammation mediator, cPLA2, contributes to obesity by inducing the infiltration of neutrophil and adipogenesis [17, 23]. On the other hand, LPS from gut microbiota was reported as linker between inflammation and high-fat diet-induced metabolic syndrome [17]. However, whether LPS directly regulated proliferation and adipogenesis of preadipocytes via promoting cPLA2 gene expression and the related molecular mechanisms were less studied. Here we reported that LPS increased mRNA and protein expression of cPLA2 in preadipocytes. Blockage the activation of JAK2, STAT3, STAT5 or AMPK significantly attenuated LPS-induced cPLA2 expression together with phosphorylation of JAK2, STAT3, STAT5 and AMPK, separately. Inhibition of JAK2 also reduced phosphorylation level of STAT3 and STAT5 in LPS-treated preadipocytes, suggesting the upstream role of JAK2 on mediating STAT3 and STAT5 activation. Moreover, LPS-stimulated DNA synthesis, increased cell numbers and cell viability were attenuated by AACOCF3, AG490 or BML-275. Attenuation JAK2/STAT or AMPK-dependent cPLA2 expression reduced LPS-mediated adipogenesis of preadipocytes. These results indicated that LPS increased preadipocytes proliferation and adipogenesis via JAK/STAT and AMPK-dependent cPLA2 expression (Fig. 7).
Several studies reported the correlation of gut microbial composition and metabolic syndromes including obesity [31]. Microbiota-related LPS played roles in linking and regulating the inflammation and metabolic outcomes [17]. The serum levels of LPS binding protein, LPB, were high in obese children than in nonobese controls [32]. LPS induced mRNA expression of cytokines and chemokines in stromal vascular (SV) cells including preadipocytes [33]. It is also been proved that LPS promoted the activation of immune cells in adipose tissue [34, 35]. However, the action of LPS directly on preadipocytes was less addressed. Here we reported that LPS directly increased proliferation and differentiation of preadipocytes. Moreover, LPS promoted cPLA2 expression in preadipocytes, and attenuation of cPLA2 abolished LPS-mediated proliferation and differentiation of preadipocytes, indicating that LPS accelerated increase of adipocytes numbers via up-regulating cPLA2 gene. These results were supported by the finding that increased adipogenesis in LPS-injected adipose tissue [36], and similar to the finding that LPS increased the expression of cytokine mRNA in preadipocytes [33]. And resembled to the finding that increased activation of inflammasome pathway in obesity of children and adolescents [37].
Activation of JAK/STAT participated in various physiological processes such as development and inflammation. It is reposted that knock down the expression of JAK2 and STAT3 attenuated adipogenesis [38, 39]. Dominant negative of STAT5 reduced adipogenesis of 3T3-L1 cells [40]. Here we found that inhibition of JAK2 reduced LPS-increased proliferation and adipogenesis of preadipocytes. LPS enhanced phosphorylation of STAT3 and STAT5 in a JAK2-dependent manner in 3T3-L1 cells. Reduced activation of JAK2/STAT pathway abolished LPS-regulated cPLA2 expression. These data revealed that LPS involved in pathologies of obesity via upregulated proliferation and adipogenesis of preadipocytes in a JAK2/STAT-dependent cPLA2 expression manner. It is found that knock out adipocyte JAK2 expression increased adiposity in both male and female mice via reducing lipolysis [39]. Constitutive activated STAT3 is found to reverse the development of brown adipose tissue in Tyk2(-/-) mice [41]. Specific deletion of STAT5 in adipocytes impaired lipid mobilization via transcription inactivation of adipose triglyceride lipase expression [42]. After affecting proliferation and differentiation of preadipocytes, whether LPS regulated lipolysis and development of brown adipose tissue will be studied in the future.
The roles of activated AMPK in obesity were controversial. It is reported that AMPK activators cannot increase the expression of p21 together with compound C-upregulated preadipocytes expansion [43]. Activation of AMPK contributed to germacrone-reduced adipogenesis [44]. It seems that the activation of AMPK in obesity was differentially regulated in response to different treatments. Here we found that pretreatment of AMPK inhibitor attenuated LPS-increased cPLA2 expression together with proliferation and adipogenesis of preadipocytes. Activation of AMPK contributed to increased numbers of preadipocyte and adipocyte in response to LPS stimulation. This is differ to what found on EGCG-stimulated preadipocytes that inhibitor of AMPK cannot reverse the proliferative effects of insulin growth factor [20]. However, ursolic acid-inhibited adipogenesis was reversed by AMPK siRNA [21]. High glucose promoted lipid accumulation of 3T3-L1 cells via substance P-activated AMPK [45]. Activation of AMPK may differently contribute to proliferation and adipogenesis of preadpipocytes.
AMPK is reported as an energy sensor and regulator of metabolism, with the suppression of immune and inflammatory processes [46]. The inhibition effects of AMPK on cytokine-activated JAK/STAT had been proved in human fibrosarcoma cells and adipocytes [47, 48]. Thus, we further demonstrated the relationship of AMPK and JAK2 in LPS-stimulated preadipocytes. The usage of AMPK siRNA reduced LPS-regulated AMPK phosphorylation but did not attenuate JAK2 activation. On the other hand, knockdown protein expression of JAK2 did not reduce LPS-regulated AMPK phosphorylation. These results suggested the independence of activated AMPK and JAK2/STAT pathway in LPS-treated preadipocytes. This is resemble to the effects of LPS on the triple-negative breast cancer cell lines, MDA-MB-231 that both AMPK and JAK2 are activated [49]. Moreover, adipocyte-derived hormone leptin is also reported to activate both AMPK and JAK2 in hepatic cells and hypothalamus [50, 51]. Whether LPS facilitated the differentiation of preadipocytes via regulating leptin expression will be elucidated further.
Collectively, based on the literature and our findings, Fig. 7 showed a model that LPS facilitated proliferation and adipogenesis or preadipocytes via up-regulated cPLA2 expression. Activation of JAK/STAT or AMPK pathway were necessary for the expression of cPLA2. Inhibition of cPLA2 reduced LPS-mediated proliferation and adipogenesis of preadipocytes. Similarly, decreased activation of JAK/STAT or AMPK pathway attenuated LPS-stimulated cPLA2 expression together with proliferation and adipogenesis of preaidpocytes. The mechanisms of LPS-stimulated cPLA2 expression may be a link between bacteria and obesity, suggesting novel strategies for treatment.
Acknowledgements
This work was supported by grants 102-CGH-FJU-02 from Cathay General Hospital, Taiwan; 9991A01 from Fu Jen Catholic University Research Foundation, Taiwan; and NSC 102-2320-B-030-001 from Ministry of Science and Technology, Taiwan.
Author Contributions
Chao-Chien Chang, Kee-Chin Sia, and Jia-Feng Chang performed the experiments; Chao-Chien Chang, Jia-Feng Chang, Chia-Mo Lin, and Wei-Ning Lin carried out the study design; Kee-Chin Sia, Chuen-Mao Yang and Kuo-Yang Huang help the set up of transfection assay. Wei-Ning Lin and Chao-Chien Chang wrote the manuscript. All authors approved the final version of the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132:2087-102
2. Bonomini F, Rodella LF, Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015;6:109-20
3. Wang K, Cao P, Wang H, Tang Z, Wang N, Wang J. et al. Chronic administration of Angelica sinensis polysaccharide effectively improves fatty liver and glucose homeostasis in high-fat diet-fed mice. Scientific reports. 2016;6:26229
4. Otto TC, Lane MD. Adipose development: from stem cell to adipocyte. Critical reviews in biochemistry and molecular biology. 2005;40:229-42
5. Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. The biology of white adipocyte proliferation. Obes Rev. 2001;2:239-54
6. Muir LA, Neeley CK, Meyer KA, Baker NA, Brosius AM, Washabaugh AR. et al. Adipose tissue fibrosis, hypertrophy, and hyperplasia: Correlations with diabetes in human obesity. Obesity (Silver Spring, Md). 2016;24:597-605
7. Stewart A, Guan H, Yang K. BMP-3 promotes mesenchymal stem cell proliferation through the TGF-beta/activin signaling pathway. Journal of cellular physiology. 2010;223:658-66
8. Yan A, Avraham T, Zampell JC, Haviv YS, Weitman E, Mehrara BJ. Adipose-derived stem cells promote lymphangiogenesis in response to VEGF-C stimulation or TGF-beta1 inhibition. Future oncology (London, England). 2011;7:1457-73
9. Xing H, Northrop JP, Grove JR, Kilpatrick KE, Su JL, Ringold GM. TNF alpha-mediated inhibition and reversal of adipocyte differentiation is accompanied by suppressed expression of PPARgamma without effects on Pref-1 expression. Endocrinology. 1997;138:2776-83
10. Wang X, Huang M, Wang Y. The effect of insulin, TNFalpha and DHA on the proliferation, differentiation and lipolysis of preadipocytes isolated from large yellow croaker (Pseudosciaena Crocea R.). PLoS One. 2012;7:e48069
11. Levine JA, Jensen MD, Eberhardt NL, O'Brien T. Adipocyte macrophage colony-stimulating factor is a mediator of adipose tissue growth. The Journal of clinical investigation. 1998;101:1557-64
12. Saint-Marc P, Kozak LP, Ailhaud G, Darimont C, Negrel R. Angiotensin II as a trophic factor of white adipose tissue: stimulation of adipose cell formation. Endocrinology. 2001;142:487-92
13. Janke J, Engeli S, Gorzelniak K, Luft FC, Sharma AM. Mature adipocytes inhibit in vitro differentiation of human preadipocytes via angiotensin type 1 receptors. Diabetes. 2002;51:1699-707
14. Hebert TL, Wu X, Yu G, Goh BC, Halvorsen YD, Wang Z. et al. Culture effects of epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) on cryopreserved human adipose-derived stromal/stem cell proliferation and adipogenesis. Journal of tissue engineering and regenerative medicine. 2009;3:553-61
15. Yamashiro H, Inamoto T, Yagi M, Ueno M, Kato H, Takeuchi M. et al. Efficient proliferation and adipose differentiation of human adipose tissue-derived vascular stromal cells transfected with basic fibroblast growth factor gene. Tissue engineering. 2003;9:881-92
16. Sethi JK. Activatin' human adipose progenitors in obesity. Diabetes. 2010;59:2354-7
17. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D. et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761-72
18. Pappo I, Becovier H, Berry EM, Freund HR. Polymyxin B reduces cecal flora, TNF production and hepatic steatosis during total parenteral nutrition in the rat. The Journal of surgical research. 1991;51:106-12
19. Nakarai H, Yamashita A, Nagayasu S, Iwashita M, Kumamoto S, Ohyama H. et al. Adipocyte-macrophage interaction may mediate LPS-induced low-grade inflammation: potential link with metabolic complications. Innate immunity. 2012;18:164-70
20. Ku HC, Liu HS, Hung PF, Chen CL, Liu HC, Chang HH. et al. Green tea (-)-epigallocatechin gallate inhibits IGF-I and IGF-II stimulation of 3T3-L1 preadipocyte mitogenesis via the 67-kDa laminin receptor, but not AMP-activated protein kinase pathway. Molecular nutrition & food research. 2012;56:580-92
21. He Y, Li Y, Zhao T, Wang Y, Sun C. Ursolic acid inhibits adipogenesis in 3T3-L1 adipocytes through LKB1/AMPK pathway. PLoS One. 2013;8:e70135
22. Hadad N, Burgazliev O, Elgazar-Carmon V, Solomonov Y, Wueest S, Item F. et al. Induction of cytosolic phospholipase a2alpha is required for adipose neutrophil infiltration and hepatic insulin resistance early in the course of high-fat feeding. Diabetes. 2013;62:3053-63
23. Pena L, Meana C, Astudillo AM, Lorden G, Valdearcos M, Sato H. et al. Critical role for cytosolic group IVA phospholipase A2 in early adipocyte differentiation and obesity. Biochim Biophys Acta. 2016;1861:1083-95
24. Yang CM, Lee IT, Chi PL, Cheng SE, Hsiao LD, Hsu CK. TNF-alpha induces cytosolic phospholipase A2 expression via Jak2/PDGFR-dependent Elk-1/p300 activation in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2014;306:L543-51
25. Zhu X, Jacobs B, Boetticher E, Myou S, Meliton A, Sano H. et al. IL-5-induced integrin adhesion of human eosinophils caused by ERK1/2-mediated activation of cPLA2. J Leukoc Biol. 2002;72:1046-53
26. Yellaturu CR, Rao GN. Cytosolic phospholipase A2 is an effector of Jak/STAT signaling and is involved in platelet-derived growth factor BB-induced growth in vascular smooth muscle cells. J Biol Chem. 2003;278:9986-92
27. Choi HI, Chung KJ, Yang HY, Ren L, Sohn S, Kim PR. et al. Peroxiredoxin V selectively regulates IL-6 production by modulating the Jak2-Stat5 pathway. Free radical biology & medicine. 2013;65:270-9
28. Kamigaki M, Hide I, Yanase Y, Shiraki H, Harada K, Tanaka Y. et al. The Toll-like receptor 4-activated neuroprotective microglia subpopulation survives via granulocyte macrophage colony-stimulating factor and JAK2/STAT5 signaling. Neurochem Int. 2016;93:82-94
29. Hsu PS, Wu CS, Chang JF, Lin WN. Leptin Promotes cPLA(2) Gene Expression through Activation of the MAPK/NF-kappaB/p300 Cascade. International journal of molecular sciences. 2015;16:27640-58
30. Chen HM, Yang CM, Chang JF, Wu CS, Sia KC, Lin WN. AdipoR-increased intracellular ROS promotes cPLA2 and COX-2 expressions via activation of PKC and p300 in adiponectin-stimulated human alveolar type II cells. Am J Physiol Lung Cell Mol Physiol. 2016;311:L255-69
31. Boulange CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome medicine. 2016;8:42
32. Kheirandish-Gozal L, Peris E, Wang Y, Tamae Kakazu M, Khalyfa A, Carreras A. et al. Lipopolysaccharide-binding protein plasma levels in children: effects of obstructive sleep apnea and obesity. J Clin Endocrinol Metab. 2014;99:656-63
33. Chung S, Lapoint K, Martinez K, Kennedy A, Boysen Sandberg M, McIntosh MK. Preadipocytes mediate lipopolysaccharide-induced inflammation and insulin resistance in primary cultures of newly differentiated human adipocytes. Endocrinology. 2006;147:5340-51
34. Castoldi A, Naffah de Souza C, Camara NO, Moraes-Vieira PM. The Macrophage Switch in Obesity Development. Frontiers in immunology. 2015;6:637
35. Huh JY, Park YJ, Ham M, Kim JB. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Molecules and cells. 2014;37:365-71
36. Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV. et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20:103-18
37. Rainone V, Schneider L, Saulle I, Ricci C, Biasin M, Al-Daghri NM. et al. Upregulation of inflammasome activity and increased gut permeability are associated with obesity in children and adolescents. International journal of obesity (2005). 2016;40:1026-33
38. Deng J, Hua K, Caveney EJ, Takahashi N, Harp JB. Protein inhibitor of activated STAT3 inhibits adipogenic gene expression. Biochemical and biophysical research communications. 2006;339:923-31
39. Richard AJ, Stephens JM. The role of JAK-STAT signaling in adipose tissue function. Biochim Biophys Acta. 2014;1842:431-9
40. Nanbu-Wakao R, Morikawa Y, Matsumura I, Masuho Y, Muramatsu MA, Senba E. et al. Stimulation of 3T3-L1 adipogenesis by signal transducer and activator of transcription 5. Molecular endocrinology (Baltimore, Md). 2002;16:1565-76
41. Derecka M, Gornicka A, Koralov SB, Szczepanek K, Morgan M, Raje V. et al. Tyk2 and Stat3 regulate brown adipose tissue differentiation and obesity. Cell Metab. 2012;16:814-24
42. Kaltenecker D, Mueller KM, Benedikt P, Feiler U, Themanns M, Schlederer M. et al. Adipocyte STAT5 deficiency promotes adiposity and impairs lipid mobilisation in mice. Diabetologia. 2017;60:296-305
43. Nam M, Lee WH, Bae EJ, Kim SG. Compound C inhibits clonal expansion of preadipocytes by increasing p21 level irrespectively of AMPK inhibition. Archives of biochemistry and biophysics. 2008;479:74-81
44. Guo YR, Choung SY. Germacrone inhibits adipogenesis and stimulates lipolysis via the AMP-activated protein kinase signalling pathway in 3T3-L1 preadipocytes. The Journal of pharmacy and pharmacology. 2017;69:202-12
45. Dubon MJ, Byeon Y, Park KS. Substance P enhances the activation of AMPK and cellular lipid accumulation in 3T3L1 cells in response to high levels of glucose. Mol Med Rep. 2015;12:8048-54
46. Weng JR, Dokla EME, Bai LY, Chen CS, Chiu SJ, Shieh TM. A 5' AMP-Activated Protein Kinase Enzyme Activator, Compound 59, Induces Autophagy and Apoptosis in Human Oral Squamous Cell Carcinoma. Basic Clin Pharmacol Toxicol. 2018
47. Mancini SJ, White AD, Bijland S, Rutherford C, Graham D, Richter EA. et al. Activation of AMP-activated protein kinase rapidly suppresses multiple pro-inflammatory pathways in adipocytes including IL-1 receptor-associated kinase-4 phosphorylation. Molecular and cellular endocrinology. 2017;440:44-56
48. Rutherford C, Speirs C, Williams JJ, Ewart MA, Mancini SJ, Hawley SA. et al. Phosphorylation of Janus kinase 1 (JAK1) by AMP-activated protein kinase (AMPK) links energy sensing to anti-inflammatory signaling. Science signaling. 2016;9:ra109
49. Lee KH, Hsu EC, Guh JH, Yang HC, Wang D, Kulp SK. et al. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator. J Biol Chem. 2011;286:39247-58
50. Uotani S, Abe T, Yamaguchi Y. Leptin activates AMP-activated protein kinase in hepatic cells via a JAK2-dependent pathway. Biochemical and biophysical research communications. 2006;351:171-5
51. Su H, Jiang L, Carter-Su C, Rui L. Glucose enhances leptin signaling through modulation of AMPK activity. PLoS One. 2012;7:e31636
Author contact
![]() Corresponding author: Wei-Ning Lin, Ph.D. Graduate Institute of Biomedical and Pharmaceutical Science, College of Medicine, Fu Jen Catholic University, No. 510 Zhongzheng Road, Xinzhuang District, New Taipei City 242, Taiwan. TEL (02) 29053398 FAX (02) 29053412 E-Mail: 081551fju.edu.tw
Corresponding author: Wei-Ning Lin, Ph.D. Graduate Institute of Biomedical and Pharmaceutical Science, College of Medicine, Fu Jen Catholic University, No. 510 Zhongzheng Road, Xinzhuang District, New Taipei City 242, Taiwan. TEL (02) 29053398 FAX (02) 29053412 E-Mail: 081551fju.edu.tw