Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(14):1648-1657. doi:10.7150/ijms.28289 This issue Cite
Research Paper
RIPK1 Inhibition Enhances Pirarubicin Cytotoxic Efficacy through AKT-P21-dependent Pathway in Hepatocellular Carcinoma
Hechen Huang1,2,3, Tianchi Chen1,2,3, Yuan Zhou1,2,3, Lei Geng1, Tian Shen1, Lin Zhou1,2,3 ![]() , Shusen Zheng1,2,3
, Shusen Zheng1,2,3 ![]()
1. Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310003, China
2. NHFPC Key Laboratory of Combined Multi-organ Transplantation, Hangzhou 310003, China
3. Collaborative Innovation Center for Diagnosis Treatment of Infectious Diseases, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China
Received 2018-7-3; Accepted 2018-10-12; Published 2018-11-5
Abstract
Pirarubicin (THP) is a new generation cell cycle nonspecific anthracycline anticancer drug. Pirarubicin and pirarubicin-based combination therapies have been demonstrated to be effective against HCC in TACE. However, the drug resistance limits its therapeutic efficacy. Receptor-interacting protein kinase 1 (RIPK1) displays a critical role in cell death. Here we found that RIPK1 and p21 may participate in the resistance to pirarubicin. In this study, we first found that inhibition of RIPK1 significantly decreased pAKT and increased p21, accompanied by G0/G1 phase cell cycle arrest and cell anti-proliferation in pirarubicin-treated hepatocellular carcinoma cells. Moreover, phosphorylation of AKT reversed the anti-proliferative effect of RIPK1 inhibitor in HCC, which proved that RIPK1-AKT-P21-dependent pathway played a key role in pirarubicin resistance. Using a mouse xenograft model, we further found that RIPK1 inhibitor combined with pirarubicin exerted synergistic anti-tumor effect in vivo. Upon exposure to pirarubicin treatment, xenografts under RIPK1 inhibition maintained higher levels of p21 than control xenografts. In conclusion, the results in our study demonstrated that RIPK1 inhibition enhances the anti-tumor effect of pirarubicin by overcoming drug resistance. RIPK1 inhibitor might be used as an adjuvant to potentiate the inhibitory effect of pirarubicin against primary hepatocellular carcinoma.
Keywords: Primary hepatocellular carcinoma, Pirarubicin, Receptor-interacting protein kinase 1, Chemoresistance, Transcatheter arterial chemoembolization
Introduction
Primary hepatocellular carcinoma (HCC) is the second leading cause of cancer-related deaths worldwide with a prognosis of a few months' survival from the time of diagnosis [1]. The frontline treatment of HCC is resection of the primary tumor, but it is limited in patients because of the highly malignant nature of the tumor. Thus, for patients with non-resectable HCC, transcatheter arterial chemoembolization (TACE) has been used in attempts to improve the survival [2]. Among various anti-tumor drugs, pirarubicin (THP), a novel anthracycline anticancer drug with lower cardio toxicity, which is more effective and faster in cellular uptake and drug plasma clearance than doxorubicin, has been widely used in TACE, liver metastases and colorectal cancer [3-6]. A previous study by Georgiades and Sieghart showed that 50% patients with HCC did not respond to initial TACE, which depended upon tumor response and chemotherapeutic resistance [7, 8]. Thus, the exploration of possible drug combinations, whose effects are addictive or synergic, raises the possibility to overcome chemoresistance and to improve outcomes.
Receptor-interacting protein kinase 1 (RIPK1) is first reported that it exerts a core function in necroptosis which is another form of programmed cell death in development, inflammation and tissue homeostasis. Its functions are regulating downstream molecules by posttranscriptional modifications including phosphorylation and ubiquitination [9]. In the liver, RIPK1 exerts great influence on the pathogenesis and prognosis of HCC [10,11]. Genetic and biochemical proofs suggest that the expression of RIPK1 is deregulated in liver cancer and regulation of RIPK1 signaling transduction has been described as a bright strategy for HCC treatment [12]. It is also shown that RIPK1 is a molecule with ''two faces'' in both pro- and anti-carcinogenic functions [10,13]. Recently it was reported that chemotherapeutic agents such as sorafenib, neoalbaconol and tanshinone IIA have strongly associations with RIPK1-dependent pathway by regulating the therapeutic effects in HCC [14-16]. With regard to hepatocellular carcinoma chemoresistance, we hypothesized that RIPK1 may be an important factor for pirarubicin resistance.
The main purpose is to analyze how RIPK1 inhibition enhances pirarubicin anti-tumor effects in TACE and to find out which molecular pathway is interacting in pirarubicin resistance. We showed that the specific inhibition of RIPK1 signaling with siRNA or necrostatin-1 enhanced sensitivity to pirarubicin in vitro and in vivo. These data suggest that RIPK1 inhibition holds promise as a new strategy to improve TACE.
Material and methods
Cell culture and reagents
Huh7 and MHCC-97H cells were purchased from Cell Bank of Chinese Academy of Sciences and cultured in DMEM (06-1055-57-1ACS, Biological Industries) with 10% fetal bovine serum (04-002-1A, Biological Industries), penicillin (100U/mL), streptomycin (10μg/mL) at 37◦C with 5% CO2 in a humidified incubator. Pirarubicin (S1393), necrostatin-1 (S8037) and SC79 (S7863) were purchased from Selleck. RIPK1 antibody (ab72139), P21 antibody (ab109520), β-actin antibody (ab8226) and GAPDH antibody (ab8245), Rb antibody (ab181616) and pRbS780 antibody (ab173289) were purchased from abcam. AKT antibody (4691) and p-AKTSer473 antibody (9271) were purchased from Cell Signaling Technology. Goat anti-Mouse (A00160) and anti-Rabbit HRP (A00166) were purchased from GenScript.
Small Interfering RNA (siRNA) transfection
RIPK1-siRNA and control-siRNA were purchased from Ribobio. 5 × 105 Cells were cultured in 6 cm dishes for 36 h. Then, RIPK1-siRNA or control-siRNA was transfected into cells using Lipofectamine 2000 transfection reagent (11668019, Invitrogen) according to the manufacturer's instruction. Then the transfected cells were used for cell viability assay and Western blotting analysis.
Cell viability assay
Huh7 and MHCC-97H cells (normal or siRNA transfected) were cultured in 96-well plates with 5000 cells per well for 48 hours. The cell viability and IC50 of pirarubicin were assessed by CCK-8 reagent (CK04, Dojindo).
Quantitative real time PCR (RT-PCR)
Total RNA samples were extracted with Trizol (15596018, Invitrogen). A Rever Tre Ace-a-reverse transcription kit (RR047A, Takara) was performed to synthesize complementary DNA. The cDNA was quantified by qPCR using SYBR Premix Ex Taq (RR820A, Takara). The primer nucleotide sequences were as follow: RIPK1 (5'- GATTGGTGGGACGAGTTCAT -3' and 5'-TGTGTGAAGCCCAGTTTACG -3') and GAPDH (5'- GAACATCATCCCTGCCTCTACT -3' and 5'- ATTTGGCAGGTTTTTCTAGACG -3'). Light Cycler Data Analysis Version (3.1.102, Roche) was used to analyze the qPCR data. The fold change in mRNA was analyzed through relative quantification (2-∆∆Ct).
Western blotting
Cells were collected and lysed in lysis buffer (9803, Cell Signaling Technology) with phosphatase Inhibitor Cocktail (78444, Life Technologies) at 4°C. The protein levels were measured by BCA assay kit (23225, Thermo Scientific). After boiling for 10 min in loading buffer (NP0007, Invitrogen), equal amounts of protein were separated by 10% SDS-PAGE (NP0301BOX, Invitrogen) and then transferred to 0.45 μm PVDF membrane (IPVH00010, Millipore). The membrane was blocked with 5% nonfat dried milk containing 0.1% Tween-20 (TBST) at room temperature for 1 h and incubated with specific primary antibodies overnight at 4°C. The membrane was washed and then incubated with appropriate secondary antibodies (1:4000) for 1 h at room temperature. After washed, bolts were visualized by ChemiDocTM MP System (Bio-Rad).
EdU assay
Cell proliferation was measured by EdU 488 Imaging kit (C10310-3, Ribobio). 5000 cells in 96-well plates with different treatments were exposed to EdU (1:1000) for 2 h at 37°C. The cells were then fixed with 4% paraformaldehyde for 30 min, followed by addition of glycine and 0.5% Triton X-100 at room temperature. Cells were treated with Fluorescent dye-488 cocktail (Apollo®) for 30 min. The DNA were stained with Hoechst 33342 (1:100) and photographed by fluorescent microscope (Olympus).
Cells (1 × 106) in 6-well with different treatments were incubated for 2 h with EdU (1:1000). Cells were harvested, fixed and permeabilized with Triton X-100. The percentage of EdU-incorporated cells and maximum EdU-coupled fluorescence intensity were detected with EdU 488 flow cytometry kit (C10338-3, Ribobio). 10000 cells were collected for each sample by BD FACS Canto II (BD). These results were analyzed by FlowJo software.
Cell Cycle analysis
Cells (1 × 106) were cultured in 6 well plates and incubated with different treatments for 48 h. Cells were fixed with 70% ethanol for 24 h at 4°C. Cells were re-suspended in solution with 50 μg/ml propidium-iodide (CCS012A, Multi Sciences) for 20 min. 10000 cells were sorted by BD FACS Canto II (BD) and analyzed by ModFit software.
Cell apoptosis analysis
Cell apoptosis was quantified using the Annexin V/ propidium iodide (PI) detection kit (AD10, Beyotime). Cells (1×106/well) were plated in 6-well plates. After different treatments, cells were collected and incubated in 400 μl binding buffer with 5 μl Annexin V-FITC and 5 μl PI in dark for 15 min at room temperature. At least 10,000 cells were collected for each sample by BD FACS Canto II. These results were analyzed by FlowJo software.
Immunofluorescent staining
2000 cells were plated in 24-well plates and cultured for 24 h. After different treatments for 48h, cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. Cells were blocked with 10% BSA for 1 h. Then cells were incubated with primary antibody against p21 (1:200) overnight at 4°C. After washed, cells were incubated with secondary antibodies conjugated with Alexa Fluor 488 (A-11034, Invitrogen) for 1 h. The cells were counterstained with DAPI (H-1200, Vector Laboratories). Pictures were taken under a fluorescent microscope (Olympus).
HCC xenograft model
All animal procedures were performed according to the Guidelines for Ethical Conduct in the Care and Use of Nonhuman Animals in Research upon approval of the Ethics Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University. Firstly, 2×106 Huh7 cells/mice were injected subcutaneously in the lateral flank of six-week-old male nude mice. Tumor volume was calculated using the equation: Tumor size = (Width2 ×Length)/2. When the tumor volume reached about 200-400 mm3, chemotherapy was started and the tumor size was measured three times a week. Pirarubicin (20 mg/m2) or necrostatin-1 (1.65 mg/kg) was administered twice a week by percutaneous intratumor drug injection. Control mice received PBS only, according to the same schedule. At the end of the treatment, animals were sacrificed and each tumor mass was collected for laboratory analysis.
Immunohistochemistry
Tumor tissues were made into 3 μm paraffin sections and pretreated, followed by deparaffinization. After antigen retrieval, primary antibodies were applied (RIPK1, 1:200; P21, 1:100) overnight at 4°C. Slides were incubated for 30 min at 37°C with secondary antibody (PV-8000, ZSGB-BIO). HRP activity was detected using DAB+ Substrate Chromogen System (ZLI-9018, ZSGB-BIO). The sections were photographed by microscopy (Zeiss, Oberkochen, Germany).
Statistical analysis
All experiments were performed in triplicates. All statistical analyses were performed using SPSS 20 for Windows (SPSS Inc.). The unpaired Student's t test (2-tailed) was used for the comparison of measurable variants.
Results
RIPK1 inhibition time- and dose-dependently enhanced pirarubicin cytotoxic efficacy in hepatocellular carcinoma cells
To explore the pharmacodynamics of pirarubicin, a dose-time-effect study was performed in Huh7 and MHCC-97H cell lines. We initially explored the effects of RIPK1 silencing (Figure 1A). Next, to determine whether pirarubicin cytotoxic efficacy observed in cells was associated with the levels of RIPK1, we ablated endogenous RIPK1 expression by transient siRNA transfection and functionally inhibited RIPK1 protein activity by necrostatin-1 [17, 18].
RIPK1 inhibition time- and dose-dependently enhanced pirarubicin cytotoxic efficacy in Huh7 and MHCC-97H cells. Notes: (A) Transfection with RIPK1-siRNA decreased expressions of RIPK1 in cells. (B) Cells were exposed to the designated concentrations of pirarubicin or necrostatin-1 for 24 or 48 h. (C) Cells transfected with control or RIPK1-siRNA were treated by pirarubicin at the designated concentrations for 24 or 48 h. (D) Necrostatin-1 (50 μM) enhanced the anti-proliferative effect of pirarubicin (0.3 μM) at 48 h. Significant differences among different treatments are marked with different letters, * (p < 0.05), ** (p < 0.01), *** (p < 0.001), **** (p< 0.0001) and ns (not significant).
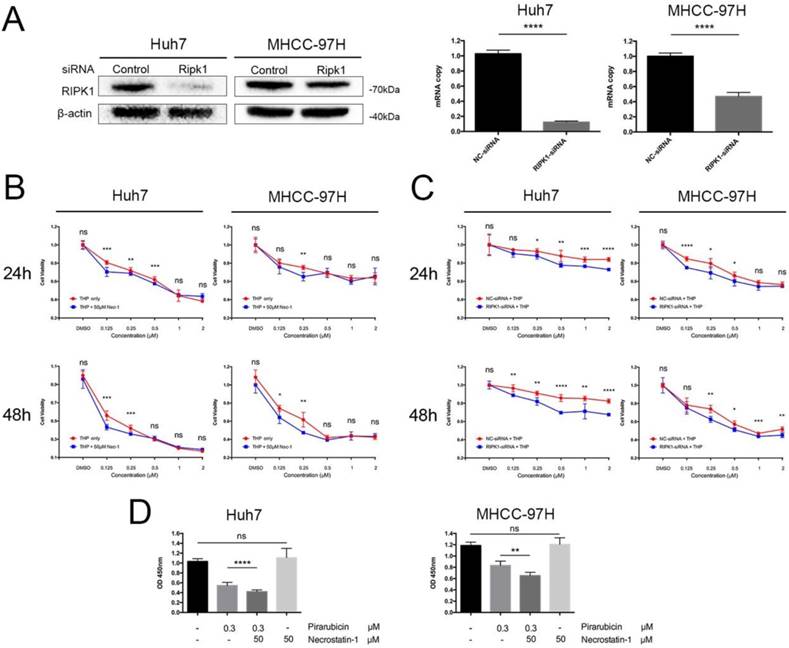
As shown in Figure 1B, C and Table 1, cell proliferation was obviously suppressed after pirarubicin treatment. The results also showed that inhibition of RIPK1 sensitized hepatocellular carcinoma cells to pirarubicin. When combined with necrostatin-1, IC50 of pirarubicin in MHCC-97H cells decrease by 21% at 24 h; Moreover, at 48 h, it decreased by 58% and 32% in Huh7 and MHCC-97H cells respectively (Figure 1B). Figure 1C showed the results of similar experiments performed with RIPK1-siRNA. IC50 of pirarubicin decreased by 89% and 6% at 24 h in Huh7 and MHCC-97H cells; Moreover, at 48 h, it decreased by 78% and 28% respectively. Inhibition of RIPK1 by necrostatin-1 or RIPK1-siRNA alone did not influence the proliferation of HCC, so the synergism between pirarubicin and RIPK1 inhibition is dominated by pirarubicin (Figure 1D). What's more, RIPK1 inhibition enhances pirarubicin anti-tumor effects, which depends on low concentrations (about 0.125-0.5 μM) of pirarubicin, indicating that hepatocellular carcinoma cells show a time- and dose-dependent sensitivity towards pirarubicin.
the IC50 of pirarubicin in different treatment group in HCC
| Cell lines | Huh7 | MHCC-97H | ||
|---|---|---|---|---|
| Treatment | IC50 24h | IC50 48h | IC50 24h | IC50 48h |
| Pirarubicin | 0.904 | 0.159 | 4.118 | 0.374 |
| Pirarubicin + Necrostatin-1 | 0.860 | 0.067 | 3.239 | 0.255 |
| NC-siRNA + Pirarubicin | 56.246 | 35.81 | 2.469 | 0.846 |
| RIPK1-siRNA + Pirarubicin | 19.947 | 7.898 | 2.323 | 0.607 |
Notes: The unit is μM.
RIPK1 inhibition enhanced the anti-proliferative effects of pirarubicin in hepatocellular carcinoma cells
Next, the present study explored the implication of RIPK1 in the therapeutic effects of pirarubicin. We first examined the apoptosis level of Huh7 and MHCC-97H cells. But neither necrostatin-1 nor low concentrations of pirarubicin, nor the combination, had any effect on apoptosis in short time (Figure S1). Furthermore, we demonstrated the role of pirarubicin or necrostatin-1 in cell cycle distribution. Pirarubicin singly caused a G2/M arrest extremely, but in combination with necrostatin-1, the percentage of G0/G1 phase was increased while that of G2/M phase was decreased (Figure 2A, S2). To further quantitatively extract the accurate kinetics of S phase and evaluate anti-proliferative effects, cells were assessed by EdU incorporation assay. Combination of pirarubicin and necrostatin-1 obviously decreased the cell number of incorporated EdU (Figure 2B). The percentage of EdU-incorporated cells and maximum EdU-coupled fluorescence intensity determined by flow cytometry indicated that necrostatin-1 indeed decreased the number of cells in S phase (Figure 2C, D and S3). These data indicated that the synergism induces cell cycle arrest at both the G0/G1 and G2/M phase, and restricts the S phase before the detection of apoptosis.
Inhibition of RIPK1 enhanced the growth-inhibitory effects of pirarubicin in Huh7 and MHCC-97H cells. Notes: (A) Pirarubicin and RIPK1 regulated the distribution of cell cycle in 48 h. Results were depicted as the percentage of G0/G1, G2/M or S phase. (B) Cell proliferation was analyzed by EdU incorporation EdU 488 imaging assay. (Scale bar, 50μm) (C) The levels of EdU-DNA in cells were detected by EdU 488 flow cytometry assay. Fluorescence intensities are displayed along the X-axis in logarithmic scale. (D) The diagrams (left panel), which displayed the counts of EdU-positive cells with different treatments, were merged into one (Red: Control; Orange: pirarubicin only; Blue: pirarubicin with necrostatin-1). Histograms (right panel) indicating proliferating cells (S phase) were depicted as the percentage of EdU incorporation. Significant differences among different treatments are marked with different letters, * (p < 0.05), ** (p < 0.01), *** (p < 0.001) and ns (not significant).
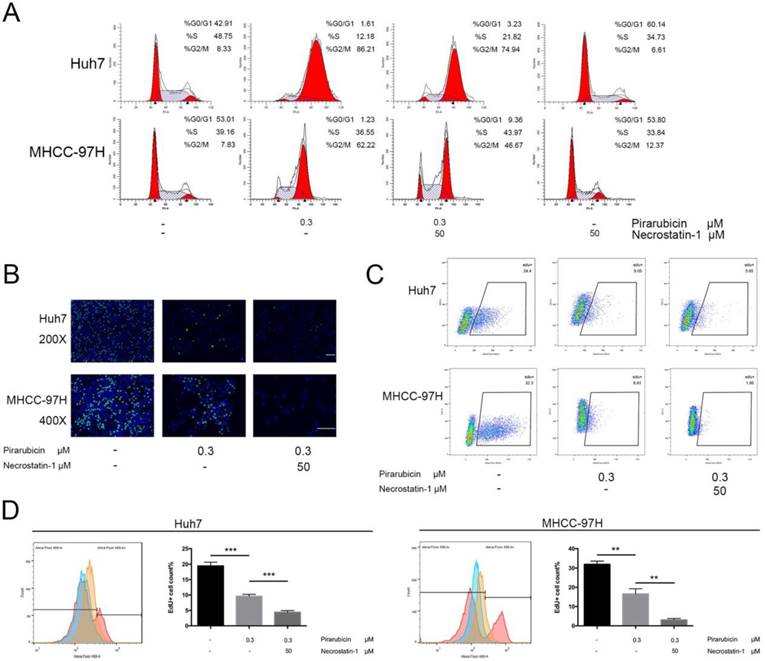
These findings show that inhibition of RIPK1 enhances the growth-inhibitory effects of pirarubicin and indicate that RIPK1 inhibition mediating G0/G1 phase arrest may play a role in pirarubicin resistance.
Combination of RIPK1 and pirarubicin leaded to down-regulation of p-AKT and up-regulation of P21 in hepatocellular carcinoma cells
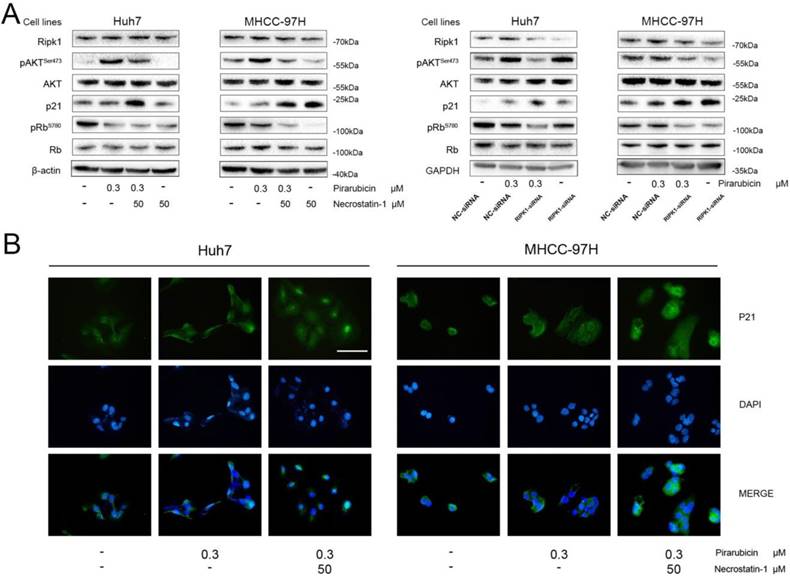
To determine how the synergism between pirarubicin and RIPK1 inhibition could regulate the cell cycle distribution, the expressions of key molecules were examined in Huh7, MHCC-97H cells. It was reported that necrostatin-1 inhibited the increase of p-AKTSer473, confirming that phosphorylation of AKT in response to TNFa/zVAD were dependent on RIPK1 in necroptosis [19,20]. These studied indicate that AKT is a downstream target of PIPK-dependent pathway. We also examined the protein status of the G0/G1 phase such as p21, Rb [21-24].
We examined AKT, which is an indicator of cell proliferation, and its activated phosphorylation status (p-AKTSer473) (Figure 3A). In this case, the pAKTSer473 levels were up-regulated in response to pirarubicin, with no change in total AKT levels. The result tallies with the report [25]. Surprisingly necrostatin-1 or RIPK1-siRNA reversed the high levels of pAKTSer473 caused by pirarubicin. At the same time, we assessed the levels of p21 by western blot and immunocytochemistry (Figure 3A, B). Pirarubicin induced few expressions of p21 in cells. What is more, the expressions of p21 were further enhanced by necrostatin-1 or RIPK1-siRNA significantly. When combined with pirarubicin, necrostatin-1 also promoted the nuclear localization of p21. Intriguingly, necrostatin-1 or RIPK1-siRNA also decreased the expression of p-Rb in cells treated with pirarubicin. (Figure 3A). These data demonstrate that the synergism between pirarubicin and RIPK1 inhibition provides a novel regulatory mechanism to restrict the S phase by specific down-regulation of p-AKTSer473 and up-regulation of p21.
RIPK1 and pirarubicin regulated p-AKT and cell cycle protein levels in Huh7 and MHCC-97H cells. Notes: (A) Cells were exposed to the designated treatments for 48 h. AKT and cell cycle proteins were measured by western blotting. β-actin and GAPDH served as loading controls. (B) Representative immunofluorescent staining showed the protein expressions and intracellular location of p21 in cells with the designated treatment. (Scale bar, 50 μm)
RIPK1 inhibition enhanced pirarubicin cytotoxic efficacy via RIPK1-AKT-P21-dependent pathway
To determine whether p-AKTSer473 is the possible bridge between p21 and RIPK1, an AKT phosphate agonist (SC79) was added to Huh7 and 97H cells treated with pirarubicin and necrostatin-1 [26,27]. Additional treatment of SC79 resulted in up-regulation of p-AKTSer473, down-regulation of p21 and a reverse of cell viability, compared with combination of pirarubicin and necrostatin-1 (Figure 4A, B).
RIPK1-AKT-P21-dependent pathway played a key role in pirarubicin resistance. Notes: (A) Huh7 and MHCC-97H cells were exposed to the designated concentrations of pirarubicin, necrostatin-1 or SC79 for 48 h. The cell viabilities were measured by CCK-8 assay. (B) The expressions of AKT and p21 were measured by western blotting. GAPDH served as a loading control. Significant differences among different treatments are marked with different letters, * (p < 0.05), ** (p < 0.01), ***(p < 0.001), **** (p< 0.0001) and ns (not significant).
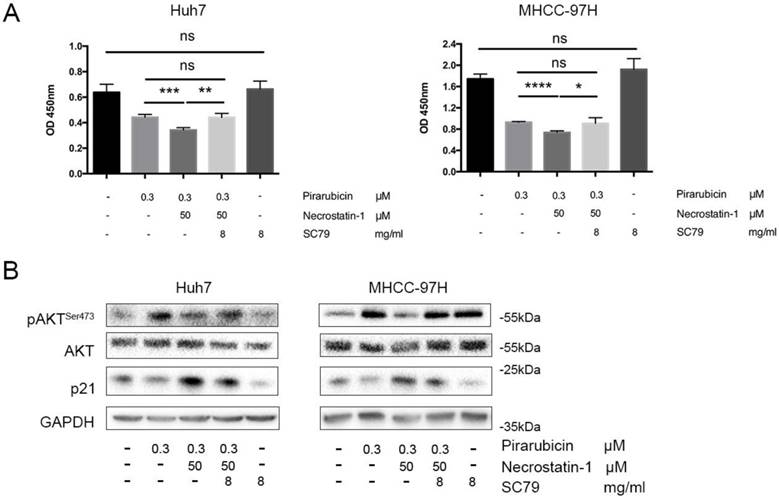
The converse experiment strengthens the above hypothesis that p21 and p-AKTSer473 are the major effectors in RIPK1-AKT-P21-dependent pathway to enhance pirarubicin sensitivity in hepatocellular carcinoma cells.
Combination of necrostatin-1 and pirarubicin exerted an antitumor effect in vivo via up-regulation of p21
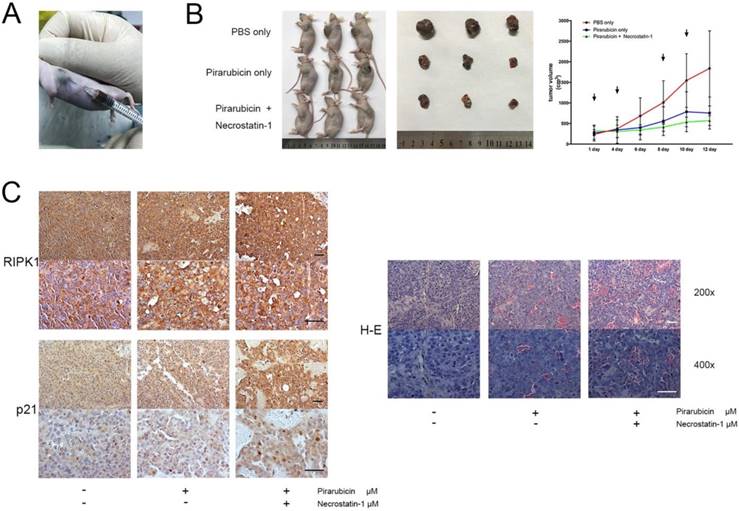
The above findings promoted us to further find out the interaction between pirarubicin and RIPK1-AKT-P21 signal pathway in vivo and to imitate the antitumor efficacy in TACE. For this purpose, a subcutaneous xenograft nude mice model was generated to investigate anti-tumor proliferation effect, and a percutaneous intratumor drug injection model was used to imitate TACE (Figure 5A) [28-30]. In our model, we showed that combination of necrostatin-1 and pirarubicin exerted a suppressive effect on the tumorigenicity of Huh7 cells, which confirmed the viewpoint that the synergism between pirarubicin and RIPK1 inhibition decelerates HCC growth in vivo (Figure 5B). Moreover, we used immunohistochemistry to assess the expressions and location of RIPK1 or p21 in xenograft mouse tumors. Combination of necrostatin-1 and pirarubicin induced necrosis much severer than other treatment groups. Compared with PBS or pirarubicin alone, combination of necrostatin-1 and pirarubicin increased the expression of p21 and promoted the nuclear localization of p21 in xenograft tumors. Neither necrostatin-1 nor pirarubicin did affect the expression and location of RIPK1. These facts further demonstrated that the joint action of pirarubicin and necrostatin-1 retarded HCC growth via anti-proliferative effect and necrosis (Figure 5C).
Pirarubicin combined with necrostatin-1 inhibited HCC xenograft growth. Notes: (A) a subcutaneous xenograft nude mice model and a percutaneous intratumor drug injection model (B) Photograph of nude mice (left panel) and photograph of dissected xenograft tumors from nude mice (middle panel) were shown. 3 mice per group. Photograph (right panel) represented the tumor volumes at the indicated days (Arrowheads denote the date of drug injection). (C) Immunohistochemical staining (left panel) and H&E staining (right panel) were shown. (Scale bar, 50 μm).
Discussion
It was shown, in TACE, that pirarubicin significantly prolongs survival of patients with liver cancer, but the tumor response was limited because of drug resistance [7,31-33]. Thus, it is great important to identify cellular signal pathways targeted to enhance sensitivity of pirarubicin, or to understand the mechanisms of pirarubicin resistance in TACE. Activation of AKT in response to cellular stress is a generalized, compensatory self-defense mechanism to escape death [25]. In our study, hepatocellular carcinoma cells likely perceive pirarubicin chemotherapy as a cellular insult. Within cells, anthracyclines have pleiotropic actions including generation of reactive oxygen species, inhibition of topoisomerase II, and induction of DNA damage. A sustained high level of Akt activity (over 24 h) was observed in breast cancer cells with doxorubicin, and a small molecular PI3K/AKT inhibitor - LY294002 potentiated cell death caused by doxorubicin [34]. Combinations of PI3K/AKT/mTOR pathway inhibitors such as perifosine, CCI-779 and RAD-001 with various types of chemotherapy have been investigated in clinical studies, but poor solubility, high toxicity and negative difference in OS have limited their clinical application [35]. In the present study, we report that inhibition of RIPK1, which is an upstream of AKT, enhances pirarubicin toxicity towards HCC cells both in vitro and in vivo.
We found that inhibition of RIPK1 changed cell cycle distribution and enhanced cell anti-proliferation inducing effect of low concentration of pirarubicin via specific down-regulation of p-AKTser473 and up-regulation of p21. p-AKTSer473 raised after exposure to pirarubicin while it returned to baseline levels because of RIPK1 inhibition. The strong activation of AKT indicates that pirarubicin might activate the cell's self- defense mechanism and resist the pirarubicin cytotoxic efficacy. In addition to being activated by drugs or reactive oxygen species, AKT can be activated by other stresses such as hypoxia, hypoglycemia and even siRNA transfection. So, we used necrostatin-1 as well as RIPK1-siRNA to demonstrate the relationship between RIPK1-dependent pathway and pirarubicin resistance in HCC. As far as p21 is concerned, high expression of p21 inhibits activities of G1/S phase cdk-cyclin complex kinases. After that, Rb protein cannot be phosphorylated and E2F cannot be released, so that the cell cycle is arrested at G0/G1 phase and DNA replication is inhibited [21,36]. It is reported that pirarubicin induced few expressions of p21 in cells, because p21 is also required to sustain G2 phase arrest caused by anthracycline anticancer drugs [37]. In addition, p-AKT acts as an inhibitor of p21 and triggers consequent cellular response. Zhou proved that blocking the AKT pathway restored the nuclear localization and cell-growth-inhibiting activity of p21 [38]. In our study, low expression or function inhibitory response of RIPK1 extremely potentiated the expressions and nuclear localization of p21 in cells treated with pirarubicin. Furthermore, combination of necrostatin-1 and pirarubicin induces down-regulation of p-AKTser473 and up-regulation of p21, and enhanced the growth-inhibitory effects of pirarubicin, whereas up-regulation of p-AKTser473 caused by additional SC79 reverses these effects, suggesting that the high levels of p21 and cell anti-proliferation are dependent on down-regulation of p-AKTSer473. Although we don't provide the underlying mechanism how RIPK1 regulates AKT phosphorylation, ours is the first report to show that RIPK1-AKT-P21-dependent pathway is involved in mediating pirarubicin resistance.
Inhibition of hepatocellular carcinoma growth is considered as a major mechanism of pirarubicin in TACE. To simulate TACE, we generated a subcutaneous xenograft nude mice model and a percutaneous intratumor drug injection model to confirm our hypothesis. We proved that combination of pirarubicin and RIPK1 inhibition resulted in slower tumor growth, as demonstrated by tumor volume and p21 staining in vivo.
The present results suggest, for the first time, that RIPK1 inhibition enhances the anti-tumor effect of pirarubicin by overcoming chemoresistance in HCC. Importantly, we show evidence that the effects of RIPK1 inhibition in pirarubicin resistance are mediated via RIPK1-AKT-P21-dependent pathway. Our data suggest that RIPK1 inhibitors may evaluate as a promising strategy, targeting RIPK1 should be preferred in TACE.
Abbreviations
IC50: half maximal inhibitory concentration; THP: Pirarubicin; Nec-1: necrostatin-1; RIPK1: Receptor-interacting protein kinase 1; NC-siRNA: negative control - siRNA; DMSO: dimethyl sulfoxide; EdU: 5'-ethynyl-2'-deoxyuridine; DAPI: 4',6-diamidino-2-phenylindole; PBS: phosphate buffered solution; H-E: hematoxylin-eosin.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by Innovative Research Groups of National Natural Science Foundation of China (No. 81721091) and National S&T Major Project (No. 2017ZX10203205). This study was also supported by the Zhejiang Provincial Natural Science Foundation of China (No. LY18H030002, No. LQ15H030003). This study was also supported by Zhejiang Provincial Public Welfare Technology Research Program (No. LGF18C100001). This study was also supported by the Fundamental Research Funds for the Central Universities.
Author contributions
Hechen Huang designed the experiments; Hechen Huang and Yuan Zhou performed the experiments; Hechen Huang analyzed the data; Tianchi Chen contributed reagents; Hechen Huang and Tianchi Chen wrote the paper; Lei Geng, Tian Shen, Lin Zhou and Shusen Zheng helped to draft the manuscript. All authors approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL. et al. Global cancer statistics, 2012. CA: A Cancer Journal for Clinicians. 2015;65:87-108
2. Lei J, Zhong J, Luo Y. et al. Response to transarterial chemoembolization may serve as selection criteria for hepatocellular carcinoma liver transplantation. Oncotarget. 2017;8:91328-91342
3. Okada M, Kudo S, Miyazaki O. et al. Antitumoral efficacy and pharmacokinetic properties of pirarubicin upon hepatic intra-arterial injection in the rabbit V x 2 tumour model. British Journal of Cancer. 1995;71:518-24
4. Tsurumi H, Hara T, Goto N. et al. A phase II study of a THP-COP regimen for the treatment of elderly patients aged 70 years or older with diffuse large B-cell lymphoma. Hematology & Oncology. 2007;25:107-14
5. Daruwalla J, Nikfarjam M, Greish K. et al. In vitro and in vivo evaluation of tumor targeting styrene-maleic acid copolymer-pirarubicin micelles: Survival improvement and inhibition of liver metastases. Cancer Science. 2010;101:1866-74
6. Daruwalla J, Greish K, Nikfarjam M. et al. Evaluation of the effect of SMA-pirarubicin micelles on colorectal cancer liver metastases and of hyperbaric oxygen in CBA mice. Journal of Drug Target. 2007;15:487-95
7. Georgiades C, Geschwind JF, Harrison N. et al. Lack of response after initial chemoembolization for hepatocellular carcinoma: does it predict failure of subsequent treatment? Radiology. 2012;265:115-23
8. Sieghart W, Hucke F, Pinter M. et al. The ART of decision making: retreatment with transarterial chemoembolization in patients with hepatocellular carcinoma. Hepatology. 2013;57:2261-73
9. Linkermann A, Green DR. Necroptosis. New England Journal of Medicine. 2014;370:455-65
10. Schneider AT, Gautheron J, Feoktistova M. et al. RIPK1 Suppresses a TRAF2-Dependent Pathway to Liver Cancer. Cancer Cell. 2017;31:94-109
11. Saeed WK, Jun DW. Necroptosis: an emerging type of cell death in liver diseases. World Journal of Gastroenterology. 2014;20:12526-32
12. Vucur M, Reisinger F, Gautheron J. et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Report. 2013;4:776-90
13. Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Molecular Cell. 2014;56:469-80
14. Ramírez-Labrada A, López-Royuela N, Jarauta V. et al. Two death pathways induced by sorafenib in myeloma cells: Puma-mediated apoptosis and necroptosis. Clinical Translational Oncology. 2015;17:121-32
15. Yu X, Deng Q, Li W. et al. Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNFalpha and ROS production. Oncotarget. 2015;6:1995-2008
16. Lin CY, Chang TW, Hsieh WH. et al. Simultaneous induction of apoptosis and necroptosis by Tanshinone IIA in human hepatocellular carcinoma HepG2 cells. Cell Death Discovery. 2016;2:16065
17. Xie T, Peng W, Liu Y. et al. Structural basis of RIP1 inhibition by necrostatins. Structure. 2013;21:493-9
18. Degterev A, Hitomi J, Germscheid M. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature Chemical Biology. 2008;4:313-21
19. Park S, Zhao D, Hatanpaa KJ. et al. RIP1 Activates PI3K-Akt via a Dual Mechanism Involving NF-KB-Mediated Inhibition of the mTOR-S6K-IRS1 Negative Feedback Loop and Down-regulation of PTEN. Cancer Research. 2009;69:4107-11
20. Liu Q, Qiu J, Liang M. et al. Akt and mTOR mediate programmed necrosis in neurons. Cell Death Disease. 2014;5:e1084
21. Harper JW, Adami GR, Wei N. et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805-16
22. Chen J, Jackson PK, Kirschner MW. et al. Separate domains of p21 involved in the inhibition of Cdk kinase and PCNA. Nature. 1995;374:386-8
23. Rössig L, Jadidi AS, Urbich C. et al. Akt-dependent phosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothelial cells. Molecular Cell Biology. 2001;21:5644-57
24. Broude EV, Swift ME, Vivo C. et al. p21(Waf1/Cip1/Sdi1) mediates retinoblastoma protein degradation. Oncogene. 2007;26:6954-8
25. West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resistance Updates. 2002;5:234-48
26. Yang F, Shi L, Liang T. et al. Anti-tumor effect of evodiamine by inducing Akt-mediated apoptosis in hepatocellular carcinoma. Biochemical and Biophysical Research Communications. 2017;485:54-61
27. Meyer K, Kwon YC, Ray RB. et al. N-terminal gelsolin fragment potentiates TRAIL mediated death in resistant hepatoma cells. Science Report. 2017;7:12803
28. Munck JN, Riggi M, Rougier P. et al. Pharmacokinetic and pharmacodynamic advantages of pirarubicin over adriamycin after intraarterial hepatic administration in the rabbit VX2 tumor model. Cancer Research. 1993;53:1550-4
29. Dara L, Liu ZX, Kaplowitz N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discovery. 2016;2:16089
30. Deutsch M, Graffeo CS. et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Disease. 2015;6:e1759
31. Munck JN, Rougier P, Chabot GG. et al. Phase I and pharmacological study of intra-arterial hepatic administration of pirarubicin in patients with advanced hepatic metastases. European Journal of Cancer. 1994;30A:289-94
32. Fallik D, Ychou M, Jacob J. et al. Hepatic arterial infusion using pirarubicin combined with systemic chemotherapy: a phase II study in patients with nonresectable liver metastases from colorectal cancer. Annals of Oncology. 2003;14:856-63
33. Oue T, Fukuzawa M, Kusafuka T. et al. Transcatheter arterial chemoembolization in the treatment of hepatoblastoma. Journal of Pediatric Surgery. 1998;33:1771-5
34. Clark AS, West K, Streicher S. et al. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Molecular Cancer Therapeutics. 2002;1:707-17
35. LoPiccolo J, Blumenthal GM, Bernstein WB. et al. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resistance Updates. 2008;11:32-50
36. Harper JW, Elledge SJ, Keyomarsi K. et al. Inhibition of cyclin-dependent kinases by p21. Molecular Biology of the Cell. 1995;6:387-400
37. Bunz F, Dutriaux A, Lengauer C. et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497-501
38. Zhou BP, Liao Y, Xia W. et al. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nature Cell Biology. 2001;3:245-52
Author contact
![]() Corresponding authors: Shusen Zheng, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, China. Email: shusenzhengedu.cn; Tel.: +86-571-87236570 Lin Zhou, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, China. Email: zhoulin99edu.cn; Tel.: +86-571-87236626
Corresponding authors: Shusen Zheng, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, China. Email: shusenzhengedu.cn; Tel.: +86-571-87236570 Lin Zhou, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, China. Email: zhoulin99edu.cn; Tel.: +86-571-87236626