Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Acknowledgements
Conflicts of Interest
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(13):1537-1546. doi:10.7150/ijms.24296 This issue Cite
Research Paper
Tumorous imaginal disc 1 (TID1) inhibits isoproterenol-induced cardiac hypertrophy and apoptosis by regulating c-terminus of hsc70-interacting protein (CHIP) mediated degradation of Gαs
Chih-Chung Feng1, Po-Hsiang Liao2,3, Hsiang-I Tsai2, Shiu-Min Cheng4, Liang-Yo Yang5, Vijaya PadmaViswanadha6, Lung-Fa Pan7,8, Ray-Jade Chen9, Jeng-Fan Lo10 ![]() *, Chih-Yang Huang2,3,11,12
*, Chih-Yang Huang2,3,11,12 ![]() *
*
1. Graduate Institute of Clinical Medical Science, China Medical University, Taichung, Taiwan
2. Graduate Institute of Basic Medical Science, China Medical University, Taichung, Taiwan
3. Medical Research Center For Exosomes and Mitochondria Related Diseases, China Medical University Hospital, Taichung, Taiwan
4. Department of Psychology, Asia University, Taichung, Taiwan
5. Department of Physiology, School of Medicine, College of Medicine, China Medical University, Taichung, Taiwan
6. Department of Biotechnology, Bharathiar University, Coimbatore-641 046, India
7. Cardiology Department of Taichung Armed Forced General Hospital, Taichung, Taiwan
8. Department of Medical Imaging and Radiological Sciences of Central Taiwan University of Science and Technology
9. Department of Surgery, School of Medicine, Taipei Medical University, Taipei, Taiwan
10. Institute of Oral Biology, National Yang-Ming University, Taipei, Taiwan
11. Graduate Institute of Chinese Medicine, China Medical University, Taichung, Taiwan
12. Department of Biological Science, Asia University, Taichung, Taiwan
*These authors contributed equally to this work.
Received 2017-12-11; Accepted 2018-7-30; Published 2018-10-20
Abstract
Dilated cardiomyopathy (DCM) is the most common form of non-ischemic cardiomyopathy. It is characterized by ventricular chamber dilation, and myocyte hypertrophy. Human tumorous imaginal disc 1 (Tid1), a chaperone protein and response to regulate number of signaling molecules in the mitochondria or cytosol. Tid1 also plays a major role in preventing DCM; however, the role of Tid1 in isoproterenol (ISO)-induced cardiac apoptosis and hypertrophy remains unclear. H9c2 cells were pretreated Tid1 before ISO-induced hypertrophy and apoptosis and then evaluated by IHC, TUNEL assay, IFC, Co-IP, and Western blot. From the IHC experiment, we found that Tid1 proteins were increased in tissues from different stages of human myocardial infarction. Using H9c2 cardiomyoblast cells we found that Tid1 was decreased by ISO treatment. However, over-expression of Tid1S suppressed NFATc3, BNP and calcineurin protein expression and inhibited NFATc3 nuclear translocation in ISO induced cardiomyoblast cells. On the other hand, Tid1S over-expression activated survival proteins p-AKTser473 and decreased caspase-3 and cytochrome c expression. We also found that overexpression of Tid1 enhanced CHIP expression, and induced CHIP to ubiquitinate Gαs, resulting in increased Gαs degradation. Our study showed that Gαs is a novel substrate of CHIP, and we also found that the Tid1-CHIP complex plays an essential role in inhibiting ISO induced cardiomyoblast hypertrophy and apoptosis.
Keywords: Tid1, H9c2, isoproterenol, hypertrophy, CHIP, Gαs
1. Introduction
Tumorous imaginal disc 1 (Tid1), also called Dnaja3 and mitochondrial chaperone Hsp40, is differentially expressed during cardiac development and pathological hypertrophy [1]. It is expressed in two forms. The Tid1 long (Tid1L) form increases apoptosis and the Tid1 Short (Tid1S) form suppresses apoptosis; however, these two isoforms differ only in their c-terminal tails [2, 3]. Experiments in mice have shown that Tid1 deficiency leads to dilated cardiomyopathy, progressive respiratory chain deficiency, and decreased mitochondrial DNA copy number, suggesting that Tid1 may have a cardiac protective effect against cardiomyopathy [1]. Tid1 functions as a co-chaperone with Hsp70 and causes increased proteasomal degradation of ErbB2 via the ubiquitination pathway [3, 4].
Ubiquitination is a post-translational modification that controls the quality of proteins through proteasomal degradation. It has been reported that Carboxyl-terminus of Hsc70 interacting protein (CHIP) acts as a ubiquitin ligase to control protein quality [5]. This CHIP was originally identified as a co-chaperone of Hsp40, Hsc70 and Hsp90 and has a tetratricopeptide repeat (TPR) motif and U-box domain. The TPR motif binds Hsc70 and Hsp90, and the U-box domain executes ubiquitin ligase activity. CHIP that was highly expressed in the heart was found to have a strong cardioprotective effect, demonstrated by inhibition of apoptosis following ischemia/reperfusion injury [5, 6].
Specific stimulation of β1-adrenergic receptor(β1-AR) by isoproterenol (ISO) induces hypertrophyand apoptosis in cardiac myocytes in vitro and in vivo [7, 8]. β1-adrenergic receptor signals through a stimulatory G protein (Gs) that activates adenylyl cyclase (AC) via the α-subunit (Gαs) and thereby induces the formation of cAMP and the activation of protein kinase A [9]. Specifically, β1-AR and β2-AR coupled to Gαs exert a pro-apoptotic action, while β2-AR coupled to Gi exerts an anti-apoptotic action [10]. Despite these findings, the physiological expression of Tid1 in ISO-induced myocardial apoptosis remains unclear. In the present study, we found that Tid1S overexpression induced CHIP to ubiquitinate Gαs to block ISO-induced hypertrophy and apoptosis in H9c2 cells.
2. Materials and Methods
2.1 Human cardiovascular tissue microarray
The tissue microarray (Provitro, Berlin, Germany) comprises heart specimens of normal tissue (n=10), acute infarction (n=20), granulation tissue (n=10) and myocardial scar (n=10). These tissues were obtained from females and males, with an age range from 25 to 84 years old. Infarct locations included anterior, posterior and septal parts. Normal tissues included both left and right ventricles.
2.2 Experimental animals
This study used 8-week old male spontaneously hypertensive rats (SHR) and Wistar Kyoto (WKY) rats that were purchased from BioLASCO Taiwan Co., Ltd., (Taipei, Taiwan) and performed in accordance with the Guide for the Care and Use of Laboratory Animals under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of China Medical University, Taichung, Taiwan (No.102-71-N and 104-222-N). The rats were kept in a constant temperature (22°C) with humidity of 60%. Animals were fed and tap water under a 12-h light/dark cycle. Rats were sacrificed and collected heart tissues immediately or stored at -80 °C until further use.
2.3 Cell culture and transient transfection
H9c2 cells (Rat embryonic cardiac myoblast; ATCC, VA, USA) were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Sigma-Aldrich, MO, USA) supplemented with 10% CCS (HyClone, Thermo, CA, USA), 2 mM glutamine, 1 mM pyruvate, 100 U/ml penicillin and 100 mg/ml streptomycin in humidified air (5% CO2) at 37°C. The plasmid containing the Tid1 gene was gifted from Dr. Jeng-Fan Lo, National Yang-Ming University, Taiwan. Full length pRK5-HA-Tid1S Wild type (Tid1S-WT) and pRK5-HA-Tid1S Mutant form (Tid1Su) which impaired J domain by mutating one amino acid residue within the J domain that is critical for the interaction of Tid1 with Hsp 70 proteins. In this study, H9c2 cells were grown to 80% confluence on the day of transfection. The plasmid and siRNA transfected for 24 hours using PureFection™ Nanotechnology-based Transfection Reagent (System Biosciences, CA, USA). The siRNA was purchased from Sigma (St. Louis, MO, USA), siRNA were against the following mRNA sequences: 5'-GAGAUAUCCCUGACUACUU-3'.
2.4 Western blot
Cells were lysed in 50 mM Tris-base (pH 7.4), 0.5 M NaCl, 1 M ethylenediaminetetraacetic acid (EDTA), 1 mM beta-mercaptoethanol (β-ME), 1% NP-40, 10% glycerol, IGEPAL CA-630 (Sigma-Aldrich, Missouri, USA) and protease inhibitor cocktail tablets (Roche, Mannheim, Germany) for 30 min and centrifuged at 12,000 rpm for 30 min. Proteins were separated by 8% to 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto a PVDF membrane (Millipore, MA, USA). Nonspecific protein binding was blocked using Tris-buffered saline Tween-20 (TBS-T) containing 5% skim-milk for 1 hour. Membranes were incubated with primary antibodies against Tid1 S/L, CHIP, Ubiquitin, p-NFATc3, NFATc3, BNP, ANP, cytochrome c and β-actin (Santa Cruz, CA, USA), HA (Abcam, MA, USA), calcineurin, Bcl-2 (BD bioscience, MA, USA), p-PI3K, pS473-Akt and cleaved Caspase-9 (Cell Signaling, MA, USA) at 4°C overnight. Following primary antibody incubations, membranes were incubated with horseradish peroxidase-linked secondary antibodies (1:2000) (anti-rabbit, anti-mouse, or anti-goat IgG). Protein expression was analyzed using a LAS 3000 imaging system (FUJIFILM, Tokyo, Japan).
2.5 Actin staining
Following treatments, cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and blocked with 5% skim-milk for 10 min. Cells were then washed with PBS and incubated with Rhodamine-phalloidin for 15 min, and then, nuclei were stained with DAPI for 15 min. Cells were analyzed and photographed using a fluorescence microscope, and the increasing cell area and intracellular actin polymerization were measured by Image J software. Three independent experiments were then averaged and statistically analyzed.
2.6 TUNEL assay
Cell apoptosis was detected by in situ terminal deoxynucleotide transferase-mediated dUTP nick end-labeling (TUNEL) using the In Situ Cell Death Detection Kit, Fluorescein (Roche, Mannheim, Germany), as described in the manufacturer's protocol. Cells with TUNEL-positive nuclei (green) were detected by fluorescence microscopy (Olympus, Tokyo, Japans). Using Image J software, the TUNEL positive cells were counted and analyzed statistically.
2.7 Co-immunoprecipitation (Co-IP)
H9c2 cells were lysed in 1.5 mM MgCl2, 1% Triton X-100, 50 mM pH 7.6 HEPES, 1 mM EDTA, 150 mMNaCl, 10% glycerol, 1 mM NaVO3, 10 mMNaF, 10 mM β-glycerol-phosphate and protease inhibitor cocktail tablets (Roche) for 30 min and centrifuged at 12,000 rpm for 30 min. The supernatants were collected and quantified by Bradford assay (Bio-Rad, CA, USA) for Co-IP analysis. Samples containing equal amounts of protein (700 μg) were incubated overnight with 20 µl of Protein G PLUS-Agarose Immunoprecipitation Reagent (Santa Cruz) and 5 μg of primary antibodies at 4 °C on a rotating wheel. Samples were centrifuged at 2,500 rpm for 5 min at 4° C. The precipitated protein G beads were washed and analyzed by immunoblotting.
2.8 Statistical analysis
All data were expressed as the means ± SD. The results were analyzed by Student's t-tests and one-way analysis of variance (ANOVA) to compare between two group and multiple groups, respectively. Significant differences (p<0.05) between the means of control and treatment groups were analyzed. All statistical procedures were performed with GraphPadPrism Ver 5.0 software (GraphPad Software).
3. Results
3.1 Expression pattern of Tid1 and CHIP
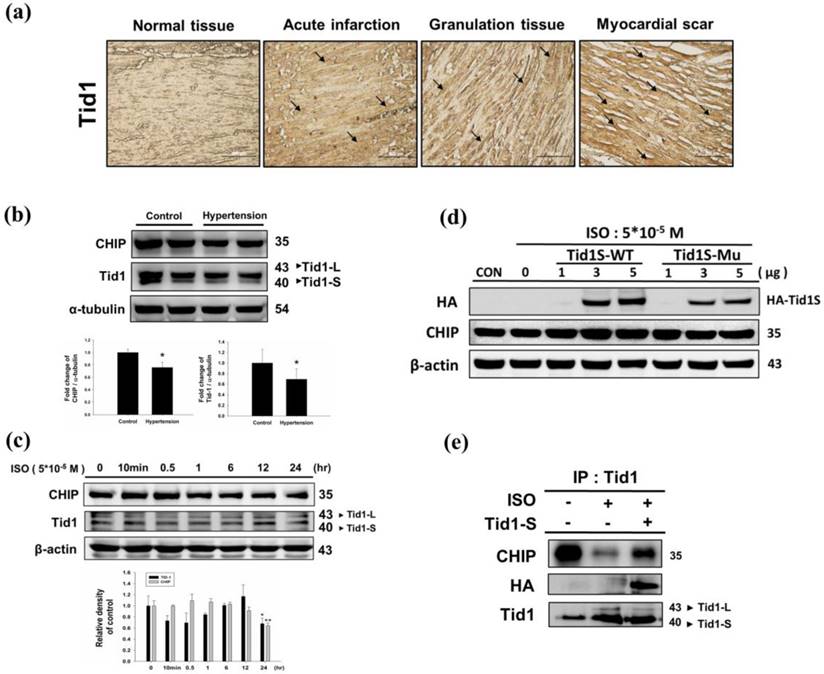
We first analyzed Tid1 expression in diseased heart by evaluating the IHC staining patterns in specimens from different stages of myocardial infarction. Tid1 staining was weaker in normal tissue when compared to acute, granulation and myocardial scar tissue samples (Fig. 1a). Next, we evaluated Tid1 and CHIP expression in the hearts of hypertension induced hypertrophy animal models. We found that the SHR rats showed decreased Tid1 and CHIP expression levels compared with the control group (Fig. 1b). To confirm this data, H9c2 cells were treated with ISO for 24 hours to mimic the hypertrophy model. As shown in Fig. 1c, ISO treatment for 10 min and 30 min increased CHIP expression. However, after 30 min of treatment, CHIP expression declined gradually in a time-dependent manner. ISO treatment for 10 min, 30 min, 1 hour and 6 hours suppressed Tid1 expression. Next, we over expressed Tid1S Wild type (Tid1S-WT) and its mutant form (Tid1Su) to determine relation of Tid1 and CHIP. We found that over expression of the Tid1S-WT form increased CHIP expression, but the mutant form was unable to increase CHIP levels (Fig. 1d). This was further confirmed by Co-IP data. As shown in Fig. 1e, ISO treatment decreased the binding interaction between CHIP and Tid1 (Fig. 1e). However, Tid1S over expression increased CHIP binding with Tid1. These data suggest that ISO inhibited Tid1 and CHIP expression and this inhibition was overcome by overexpressing Tid1S.
Expression Tid1 and CHIP in different stage of myocardial infarction tissues and H9c2 cardiomyoblast cells treated with ISO. (a) Immunohistochemical analysis, with the indicated antibodies, of serial sections of representative lesions. Tid1 accumulations were increased with disease progression visualized dark-brown color which indicated with arrows. Panels: a normal tissue (NT, n=10), an acute infraction (AI, n=10), a granulation tissue (GT, n=10) and a myocardial scar (MS, n=10). Final magnifications: × 200 (bar, 200 μm). (b) The expression of CHIP and Tid1 in hypertension models were measured via immunoblotting. Quantification of the results is shown blow from three independent experiments; mean ± S.D., * P < 0.05. (c) H9c2 cells were treated with different dosages of ISO (5*10-5 M) in serum-free medium for 24 hours were measured via immunoblotting. Quantification of the results is shown blow from three independent experiments; mean ± S.D., * P < 0.05, ** P < 0.01. (d) H9c2 cells were pretreated with ISO (5*10-5 M) in serum-free medium for 24 hours, and then transfected with different amounts of Tid1S-WT (1, 3 and 5μg) and Tid1Su plasmid (1, 3 and 5μg) for 24 hours. Cell was harvest and analyzed via immunoblotting. (e) H9c2 cells were pretreated with ISO (5*10-5 M) then transfected with Tid1S-WT plasmid and harvested for immunoprecipitation with anti Tid1 antibody, and the samples were analyzed via immunoblotting.
3.2 Isoproterenol treatment induces hypertrophy and apoptosis in H9c2 cardiomyoblast cells
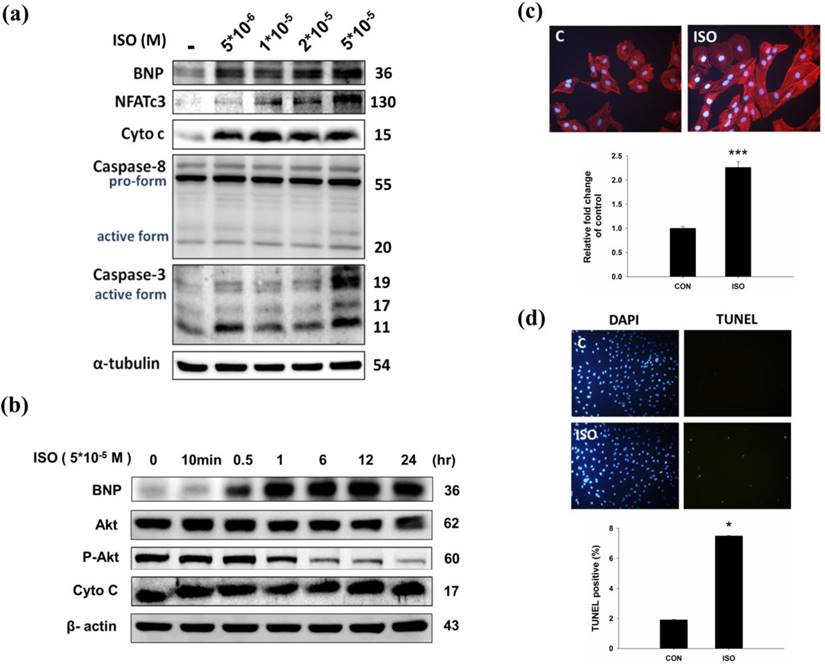
To confirm the effect of ISO in H9c2 cardiomyoblast cells, H9c2 cells were treated with ISO and then analyzed for hypertrophy and apoptosis markers. Western blotting results showed that ISO treatment increased BNP, cytochrome C, and caspase 3 expression (Fig.2a). Similarly, ISO treatment decreased phosphorylated AKTser473 expression in a dose-dependent manner (Fig. 2b). As shown in Fig 2c, ISO treatment increased NFATc3 and caspase 3 expression in dose-dependent manner. We further confirmed the effect of ISO by analyzing the cell surface area, and we found that ISO treatment increased the cell surface area compared with the control cells (Fig. 2c). A TUNEL assay was used to confirm the augmented apoptosis in ISO treated H9c2 cells. After ISO treatment, the TUNEL signals were increased (Fig. 2d). These data suggest that ISO treatment induced hypertrophy and apoptosis related protein expression in H9c2 cardiomyoblast cells.
Effects of ISO on in H9c2 cardiomyoblast cells. (a) H9c2 cells were pretreated with different dosages of ISO (5*10-6, 1*10-5, 2*10-5, 5*10-5 M) in serum-free medium for 24 hours and then analyzed for BNP, NFATc3, cytochrome C, caspase 8, caspase-3 expression via immunoblotting. (b) H9c2 cells were pretreated with different time-points of ISO (5*10-5 M) and then analyzed for BNP, p-AKT and cytochrome C expression via immunoblotting. (c) H9c2 cells were treated ISO for 24 hours, and then analyzed for Actin staining using phalloidin-rhodamine. Quantification of the results is shown blow from three independent experiments; mean ± S.D., *** P < 0.001. (d) H9c2 cells were treated ISO for 24 hours and TUNEL assay was performed after these treatments. The left panel showed DAPI stain nucleus in blue and the right panel represent TUNEL stained apoptotic nucleus in green. Quantification of the results is shown blow from three independent experiments; mean ± S.D., * P < 0.05.
3.3 Tid1S overexpression inhibits ISO induced hypertrophy and apoptosis
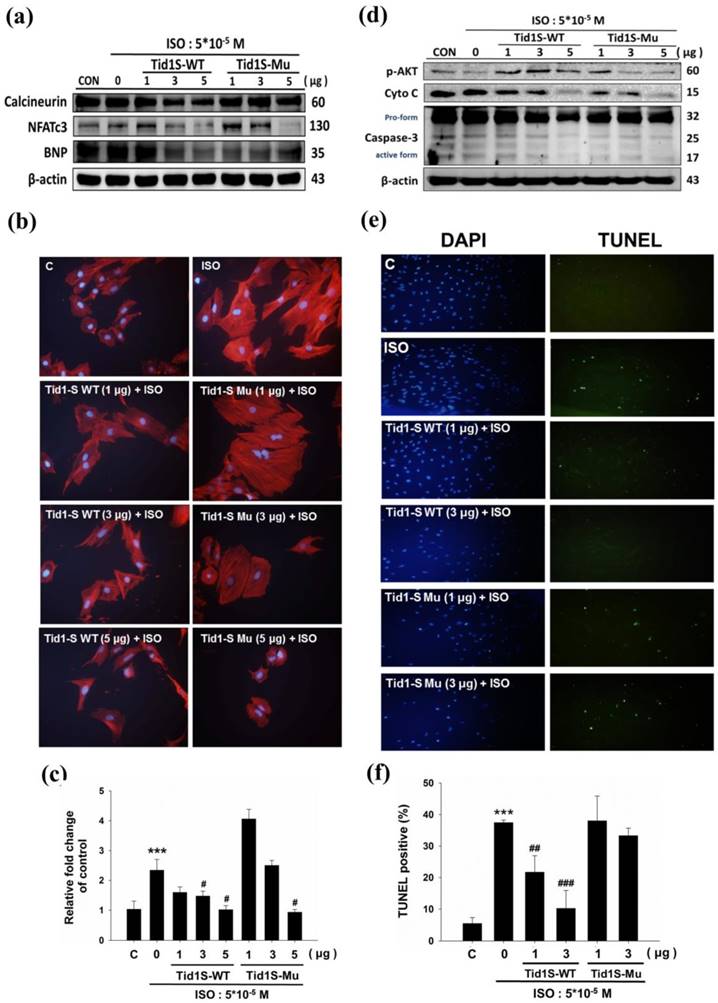
The Calcineurin/NFATc3 signaling pathway plays a crucial role in cardiac hypertrophy [11]. We therefore examined whether Tid1S prevented H9c2 cell hypertrophy in a calcineurin/NFATc3 dependent manner. To confirm this, we first overexpressed Tid1S-WT and Tid1S-Mu in cardiomyoblast cells and then treated the cells with ISO for an additional 24 hours. As demonstrated previously, ISO treatment increased expression of calcineurin and NFATc3 proteins compared with control cells. Both Tid1S-WT and Tid1Su attenuated NFATc3. Tid1S-WT overexpression down regulated calcineurin and BNP expression in ISO-treated cells (Fig. 3a), whereas Tid1S-Mu overexpression resulted in no change in calcineurin and BNP. Actin staining was further performed to confirm the role of Tid1S in reducing the cell surface area. The ISO-induced increased cell size area of H9c2 cells was decreased in Tid1S-WT transfected cells. In contrast, the Tid1S-Mu form was unable to inhibit ISO-induced cell surface area at a low transfection dose (Fig. 3b and c). These data indicate that Tid1S-WT played a preventive role in cardiac hypertrophy induced by ISO. To investigate the molecular mechanisms of cell apoptosis after Tid1S treatment, we analyzed the expression of survival proteins and apoptosis proteins by Western blotting. The results revealed that ISO inhibited p-AKT ser473 expression and increased cytochrome C and caspase-3 expression (Fig. 3d). Tid1S-WT overexpression reversed the ISO effect in H9c2 cells by increasing levels of the survival proteins p-AKTser473, and decreasing cytochrome C and caspase-3 expression. A TUNEL assay was used to confirm the apoptosis inhibitory ability of Tid1S in ISO stimulated cells. ISO increased TUNEL signals in H9c2 cells, but the signal was decreased in Tid1S-WT transfected cells (Fig. 3e and f).
Tid1S overexpression to inhibit ISO-induced hypertrophy and apoptosis in H9c2 cells.H9c2 cells transfected with Tid1S-WT (1, 3 and 5μg) and Tid1S-Mu plasmid (1, 3 and 5μg) for 24 hours before treated with ISO (5*10-5 M) for 24 hours. Total cell lysates were harvested and measured by immunoblotting. (a) Expression of Calcineurin, NFATc3 and BNP were estimated via immunoblotting. (b) Cellular morphology was observed using Actin staining and DAPI staining was used to mark the nucleus. (c) These data were obtained from three independent experiments and values represent the mean ± S.D., *** P < 0.001, represent significantly different relative to control;# P < 0.05, ## P < 0.01, represent significantly different relative to ISO. (d) Expression of p-Akt, cytochrome C and caspase-3 was analyzed using immunoblotting. (e) Cells were fixed and assayed by TUNEL test and then counter stain with DAPI (blue, nucleus). (f) Bar graphs present percentage of TUNEL positive cells. These data were obtained from three independent experiments and values represent the mean ± S.D., *** P < 0.001, represent significantly different relative to control; ## P < 0.01, ### P < 0.001, represent significantly different relative to ISO.
3.4 Tid1S overexpression inhibits ISO-induced NFATc3 nuclear translocation
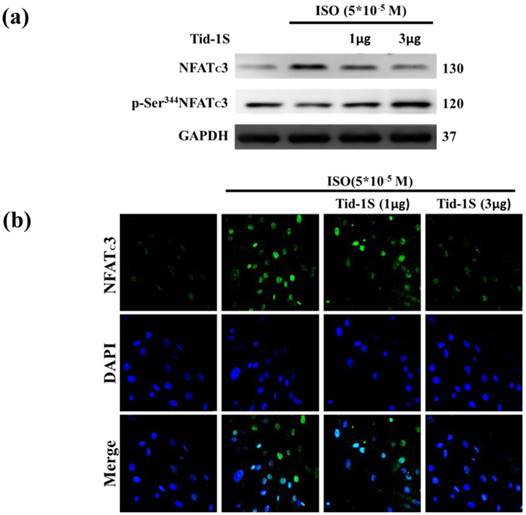
Previous observations have shown that calcineurin/NFATc3 signaling plays a critical role in regulating cardiac hypertrophic growth [12]. Therefore, we hypothesize that Tid1S overexpression may control ISO-induced NFATc3 translocation into the nucleus. We examined the distribution of NFATc3 in both cytoplasm and nucleus following Tid1S overexpression. Tid1S-WT overexpression inhibited NFATc3 nuclear translocationin ISO treated cells (Fig. 4a and b).
Tid1S overexpression inhibits ISO-induced NFATc3 nuclear translocation. H9c2 cells were transfected with different amounts of Tid1S plasmid (1 and 3μg) for 24 hours, and then treated with ISO (5*10-5 M) in serum-free medium for 24 hours. (a) Total cell lysates analyze the phospho- (cytosol) and dephospho-NFATc3 (nuclear) by immunoblotting. (b) The nuclear translocation of NFATc3 were measured using confocal microscopy. NFATc3 is shown in green fluorescence and nuclei were counter stained with DAPI (blue fluorescence).
3.5 Tid1S induced Gαs proteasome degradation via enhanced CHIP E3 ligase ability
ISO-induced hypertrophy is mediated through Gαs expression in H9c2 cells. Therefore, we examined the role of Tid1S in Gαs expression. As shown in Fig 5a, the ISO-induced increase in Gαs expression was reduced in Tid1S overexpressing cells. Next, we evaluated if the proteasome pathway is responsible for Gαs protein degradation. We first overexpressed Tid1S in H9c2 cells, then treated cells with or without ISO and the proteasome inhibitor MG132 after analyzed Gαs expression. MG132 treatment inhibited proteasomal system results ubiquitin accumulation increase Gαs expression, and Gαs protein levels were significant increase in ISO plus MG132 treatment compare with MG132 untreatment group. However, Tid1S over expression resulted in a gradual decrease Gαs expression.
Tid1S induced Gαs proteasome degradation via CHIP E3 ligase ability. (a) H9c2 cells was transfected with Tid1S plasmid (3 μg) for 24 hours before treated with ISO (5*10-5 M) and MG132 (10 μM) in serum-free medium for 24 hours. Cell lysates were harvested and analyzed for Gαs via immunoblotting. (b) H9c2 cells were harvested and estimated via immunoprecipitation and immunoblotting.
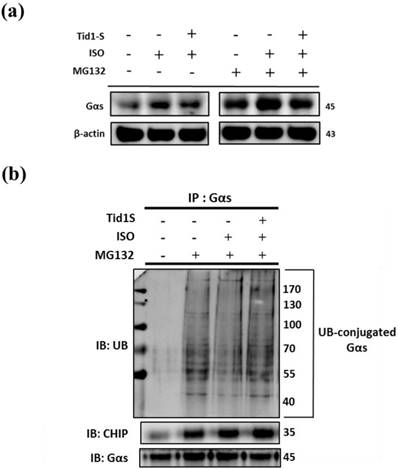
Ubiquitination is a common mechanism for protein degradation. Therefore, cells were transfected with Tid1S and then ubiquitination was analyzed by immunoprecipitation with anti-Gαs antibody, followed by immunoblotting with ubiquitin antibody. As shown in Fig 5b, MG132 significantly led to accumulation of Gαs. After ISO treatment, Gαs ubiquitination was decrease but overexpressing Tid1S increased Gαs ubiquitination after ISO treatment.
3.6 CHIP directly interacts with Gαs, whereas Tid1S facilitate CHIP-Gαs interaction in H9c2 cells
CHIP has strong cardiac protective effects through the regulation of protein quality control [6]. To address whether CHIP as a E3 ligase promotes ubiquitination and degradation of Gαs, cells were co-transfected with Tid1S and/or si-CHIP and treated with ISO or MG132. Compared to the ISO treatments, Tid1S overexpression increased CHIP binding with Gαs (Fig. 6a). In addition, blocking CHIP using si-CHIP decreased CHIP binding with Gαs, indicating that CHIP-mediated Gαs degradation was activated via Tid1S.
CHIP directly interacts with Gαs, whereas Tid1S facilitate CHIP-Gαs interaction in H9c2 cells. H9c2 cells was co-transfected with Tid1S plasmid (3 μg) and CHIP siRNA (10 nM) for 24 hours and then treated with ISO (5*10-5 M) and MG132 (10 μM) in serum-free medium for 24 hours. (a) Cell lysates were immunoprecipitated with antibodies against CHIP and analyzed CHIP and Gαs via immunoblotting. (b) Cell lysates were immunoprecipitated with antibodies against Gαs and analyzed ubiquitin, CHIP and Gαs via immunoblotting.
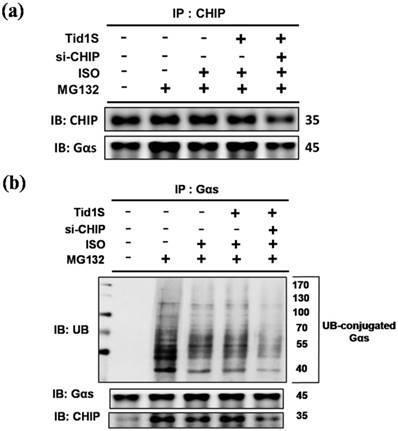
Similarly, si-CHIP markedly decreased ubiquitination of Gαs (Fig. 6b). These data suggested that ISO-induced Gαs expression was decreased by overexpressing Tid1, which binds to CHIP and activates CHIP to ubiquitinate Gαs, leading to subsequent proteosomal degradation.
4. Discussion
Several reports have found that Tid1 controls various pathways related to cell survival, cell proliferation, and stress-induced cellular responses [13]. Tid1 negatively regulates cardiomyocyte apoptosis and hypertrophy. For example, Dnaja3 or Tid1 is differentially expressed during cardiac development and pathological hypertrophy [1]. Recently, it was found that Tid1 decreases EGFR signaling by ubiquitination-mediated degradation and further reduces head and neck squamous cell carcinoma malignancy [14]. Constitutive overexpression of β-adrenergic receptors has revealed that the β1-adrenergic receptor subtype is the major mediator of pathological hypertrophy and further leads to heart failure [15]. However, the molecular mechanism by which Tid1 reduces β1-adrenergic induced hypertrophy and apoptosis remains to be addressed. The main finding of the current study was that hypertensive animals showed decreased Tid1 and CHIP expression. Hypertensive heart disease (HHD) is a major etiologic factor contributing to the appearance of heart failure [16]. Left ventricular hypertrophy and alterations in diastolic and/or systolic cardiac function were found in hypertensive patients [17]. Therefore, we used ISO to induce hypertrophy in cells. In line with these reports, our experiments showed that direct stimulation of H9c2 cells with ISO decreased both Tid1 and CHIP expression and induced hypertrophy and apoptosis. Our data also showed that β-adrenergic receptor activation disrupts direct interaction of Tid1 with CHIP. In this study, human cardiovascular tissue microarray appeared the high levels expression of Tid1 but low levels in hypertrophy animal and cell model. This result gives us a hint that the Tid1 associate to cardiac compensation between long-term and short-term disease progression.
ISO, a β-adrenergic receptor agonist, induces diffused myocardial necrosis and interstitial fibrosis in rats. Gradually, the rats presented cardiac remodeling with left ventricular hypertrophy, cardiomyopathy and heart failure-like symptoms [18]. We recently showed that ISO induced cellular calcium accumulation in cardiomyocytes by activating phospholamban (PLB) and PI3K-Akt-murine double minute 2 (MDM2) signaling cascades, which increased calcineurin protein expression, and induced myocardial cell hypertrophy and apoptosis. Consistent with this report, our current results indicate the ISO treatment induced the calcium pathway by increasing NFATC3 expression and induced hypertrophy and increased expression of apoptosis markers, including Caspase-3 and cytochrome C. Additionally, ISO treatment decreased the survival pathway by inhibiting p-AKT expression. Calcineurin is a protein phosphatase that dephosphorylates transcription factors of the NFAT (nuclear factor of activated T cells) family, which leads to their translocation to the nucleus to activate target genes. It is well established that activation of the calcineurin/NFAT pathway is sufficient for the development of cardiac hypertrophy and failure [12, 19].
Another important finding in our study was that overexpression of Tid1 reverse that ISO decreased the survival protein pAKT and reduced hypertrophy and apoptosis protein levels. Loss of Tid1 expression has been shown to be associated with human basal cell carcinoma but not with normal keratinocytes [20]. Overexpression of Tid1 reduces the tumorigenicity of breast cancer cells in nude mice and reduces anchorage-independent growth in lung adenocarcinoma cell lines [21, 22]. Similarly, in our present study ISO treatment decreased Tid1 expression; however, Tid1S overexpression blocked the ISO-induced calcineurin/NFATc3 hypertrophy pathway, reduced the apoptotic effects and increased H9c2 cell survival. From this finding, we speculate that Tid1 acts as a protective agent to prevent heart failure.
Chaperone-dependent E3 ubiquitin ligase (CHIP) forms a complex with Tid1 and promotes ErbB2 degradation [23]. Previous studies have reported that CHIP has a strong cardioprotective effect by decreasing apoptosis induced by myocardial ischemia [6]. The interaction between Tid1 and CHIP was not definitively studied in myocardial cells. In this study, we found that CHIP expression was down regulated after ISO treatment. We also found that overexpression of Tid1 increased endogenous CHIP expression and this was further confirmed by our Co-IP data. Gαs has been recognized as an important regulator of cardiomyocyte apoptosis associated with development of the failure phenotype [8, 24]. Our data showed that Tid1 overexpression degraded Gαs by activation of CHIP. Silencing of CHIP abrogate Tid1 induced Gαs degradation. We propose that CHIP is the primary E3 ligase that controls Gαs degradation in Tid1 overexpressing cells.
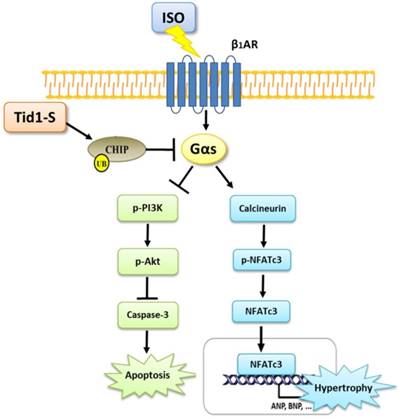
In summary, the results of this study point to a novel function of Tid1 in inhibiting ISO-induced cardiomyoblast hypertrophy and apoptosis. Tid1 induces CHIP expression and then promotes the ubiquitination and proteasomal degradation of Gαs. Furthermore, Tid1S over expression inhibited ISO-induced hypertrophy and the apoptotic pathway in H9c2 cells (Fig. 7). This study reveals that targeting Gαs for degradation may lead to protection from heart failure.
The proposed hypothesis for Tid1 enhance with CHIP-Gαs interaction and induces Gαs proteasome degradation to prevent ISO induced hypertrophy and apoptosis pathway in H9c2 cardiomyobast cells.
Acknowledgements
This study is supported in part by Taiwan Ministry of Health and Welfare Clinical Trial Center (MOHW106-TDU-B-212-113004).
Conflicts of Interest
The authors declare no conflict of interest.
References
1. Hayashi M, Imanaka-Yoshida K, Yoshida T, Wood M, Fearns C, Tatake RJ. et al. A crucial role of mitochondrial Hsp40 in preventing dilated cardiomyopathy. Nat Med. 2006;12:128-32
2. Lu B, Garrido N, Spelbrink JN, Suzuki CK. Tid1 isoforms are mitochondrial DnaJ-like chaperones with unique carboxyl termini that determine cytosolic fate. Journal of Biological Chemistry. 2006;281:13150-8
3. Syken J, De-Medina T, Munger K. TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc Natl Acad Sci U S A. 1999;96:8499-504
4. Kim SW, Chao TH, Xiang R, Lo JF, Campbell MJ, Fearns C. et al. Tid1, the human homologue of a Drosophila tumor suppressor, reduces the malignant activity of ErbB-2 in carcinoma cells. Cancer Research. 2004;64:7732-9
5. Murata S, Chiba T, Tanaka K. CHIP: a quality-control E3 ligase collaborating with molecular chaperones. International Journal of Biochemistry & Cell Biology. 2003;35:572-8
6. Zhang C, Xu Z, He XR, Michael LH, Patterson C. CHIP, a cochaperone/ubiquitin ligase that regulates protein quality control, is required for maximal cardioprotection after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2005;288:H2836-42
7. Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96:7059-64
8. Singh K, Communal C, Sawyer DB, Colucci WS. Adrenergic regulation of myocardial apoptosis. Cardiovasc Res. 2000;45:713-9
9. Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896-906
10. Amin P, Singh M, Singh K. beta-Adrenergic Receptor-Stimulated Cardiac Myocyte Apoptosis: Role of beta1 Integrins. J Signal Transduct. 2011;2011:179057
11. Tan X, Li J, Wang X, Chen N, Cai B, Wang G. et al. Tanshinone IIA protects against cardiac hypertrophy via inhibiting calcineurin/NFATc3 pathway. Int J Biol Sci. 2011;7:383-9
12. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J. et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215-28
13. Lo JF, Hayashi M, Woo-Kim S, Tian B, Huang JF, Fearns C. et al. Tid1, a cochaperone of the heat shock 70 protein and the mammalian counterpart of the Drosophila tumor suppressor l(2)tid, is critical for early embryonic development and cell survival. Mol Cell Biol. 2004;24:2226-36
14. Chen CY, Chiou SH, Huang CY, Jan CI, Lin SC, Hu WY. et al. Tid1 functions as a tumour suppressor in head and neck squamous cell carcinoma. J Pathol. 2009;219:347-55
15. Feldman DS, Carnes CA, Abraham WT, Bristow MR. Mechanisms of disease: beta-adrenergic receptors-alterations in signal transduction and pharmacogenomics in heart failure. Nat Clin Pract Cardiovasc Med. 2005;2:475-83
16. Shahbaz AU, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, McGee JE. et al. Fibrosis in hypertensive heart disease: molecular pathways and cardioprotective strategies. J Hypertens. 2010;28(Suppl 1):S25-32
17. Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568-75
18. Parveen A, Babbar R, Agarwal S, Kotwani A, Fahim M. Mechanistic clues in the cardioprotective effect of Terminalia arjuna bark extract in isoproterenol-induced chronic heart failure in rats. Cardiovascular Toxicology. 2011;11:48-57
19. Chang YM, Velmurugan BK, Kuo WW, Chen YS, Ho TJ, Tsai CT. et al. Inhibitory effect of alpinate Oxyphyllae fructus extracts on Ang II-induced cardiac pathological remodeling-related pathways in H9c2 cardiomyoblast cells. Biomedicine (Taipei). 2013;3:4
20. Canamasas I, Debes A, Natali PG, Kurzik-Dumke U. Understanding human cancer using Drosophila: Tid47, a cytosolic product of the DnaJ-like tumor suppressor gene l2Tid, is a novel molecular partner of patched related to skin cancer. J Biol Chem. 2003;278:30952-60
21. Cheng H, Cenciarelli C, Shao Z, Vidal M, Parks WP, Pagano M. et al. Human T cell leukemia virus type 1 Tax associates with a molecular chaperone complex containing hTid-1 and Hsp70. Curr Biol. 2001;11:1771-5
22. Kurzik-Dumke U, Horner M, Nicotra MR, Koslowski M, Natali PG. In vivo evidence of htid suppressive activity on ErbB-2 in breast cancers over expressing the receptor. J Transl Med. 2010;8:58
23. Jan CI, Yu CC, Hung MC, Harn HJ, Nieh S, Lee HS. et al. Tid1, CHIP and ErbB2 interactions and their prognostic implications for breast cancer patients. Journal of Pathology. 2011;225:424-37
24. Huang CY, Lee SD. Possible pathophysiology of heart failure in obesity: Cardiac apoptosis. Biomedicine (Taipei). 2012:2
Author contact
![]() Corresponding author: Chih-Yang Huang, Graduate Institute of Basic Medical Science, China Medical University, No 91, Hsueh-Shih Road, Taichung 404, Taiwan. Tel: +886-4-22053366 ext 3313; Fax: +886-4-22333641. E-mail: cyhuangcmu.edu.tw
Corresponding author: Chih-Yang Huang, Graduate Institute of Basic Medical Science, China Medical University, No 91, Hsueh-Shih Road, Taichung 404, Taiwan. Tel: +886-4-22053366 ext 3313; Fax: +886-4-22333641. E-mail: cyhuangcmu.edu.tw