Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(10):978-985. doi:10.7150/ijms.24659 This issue Cite
Research Paper
Endothelial-cell inflammation and damage by reactive oxygen species are prevented by propofol via ABCA1-mediated cholesterol efflux
Chih-Peng Hsu1#, Chih-Hung Lin2#, Chan-Yen Kuo3,4 ![]()
1. Department of Cardiology, Chang Bing Show Chwan Memorial Hospital, Changhua, Taiwan
2. Department of Internal Medicine, Cathay General Hospital, Taipei, Taiwan
3. Graduate Institute of Systems Biology and Bioinformatics, National Central University, Chungli, Taiwan
4. Department of Ophthalmology, Hsin Sheng Junior College of Medical Care and Management, Longtan, Taiwan
# The first two authors contributed equally to this work
Received 2017-12-30; Accepted 2018-5-27; Published 2018-6-14
Abstract
Background: Cholesterol efflux efficiency, reactive oxygen species, and inflammation are closely related to cardiovascular diseases. Our aim was to investigate the effect of propofol on cholesterol-loaded rat aortic endothelial cells after high-density lipoprotein treatment in vitro.
Methods and Results: The results showed that propofol promoted cholesterol efflux and ameliorated inflammation and reactive oxygen species overproduction according to the analysis of p65 nuclear translocation and a 2′,7′-dichlorofluorescin diacetate assay, respectively.
Conclusions: These results provide a possible explanation for the anti-inflammatory, antioxidant, and cholesterol efflux-promoting effects of propofol on rat aortic endothelial cells after incubation with high-density lipoprotein.
Keywords: Propofol, cholesterol efflux, reactive oxygen species, inflammation, high-density lipoprotein, rat aortic endothelial cells
Introduction
Atherosclerosis is characterised by lipid accumulation, an inflammatory response, cell death, and fibrosis in the arterial wall and is a major pathological basis for ischemic coronary heart disease, which is the leading cause of morbidity and mortality in the USA and Europe; thus, new therapeutic strategies would be warranted [1-3]. People with hyperlipidaemia are at roughly twice the risk of developing a cardiovascular disease (CVD) as compared to those with normal total cholesterol levels [4]. Therefore, a reduction in cholesterol levels by potential antiatherogenic treatments can be a useful therapeutic strategy against atherosclerosis.
Propofol (2,6-diisopropylphenol) is widely used for induction and maintenance of anaesthesia and for sedation in intensive care units [5]. The functions of propofol include anti-inflammatory effects, inhibition of production of proinflammatory cytokines, alteration of production of nitric oxide, inhibition of neutrophil function, and antioxidant properties [5, 6]. Ma et al. suggest that propofol up-regulates expression of ABCA1, ABCG1, and SR-B1 through the PPARγ/LXRα pathway in THP-1 macrophage-derived foam cells [7]. Moreover, propofol has a protective effect against myocardial ischemia-reperfusion injury in both normal rats and rats with type 2 diabetes, possibly by attenuating endothelial cell injury and by inhibiting the apoptosis of cardiomyocytes [8]. ABCA1 has been reported to mediate the secretion of cellular free cholesterol and phospholipids, thus leading to binding to an extracellular acceptor, apolipoprotein AI, to form nascent high-density lipoprotein (HDL); besides, ABCA1 is a key molecule in cholesterol homeostasis [9, 10]. Furthermore, oxidative stress is an important risk factor contributing to the pathogenesis of CVDs. Oxidative stress that results from excessive reactive oxygen species (ROS) production accounts for impaired endothelial function, a process that promotes atherosclerotic lesions or foam cell formation. Nuclear factor erythroid 2-related factor 2 (Nrf2) is involved in the protective effect against oxidative-stress-induced cardiac injury as well as in the regulation of cholesterol uptake and efflux [11]. Accumulation of excess cholesterol due to the presence of increased levels of circulating low-density lipoprotein (LDL) promotes endothelium dysfunction and activation, which is associated with increased production of pro-inflammatory cytokines, generation of ROS, and a decrease in nitric oxide levels and bioavailability [12]. The effect of propofol on oxidative-stress-induced endothelial cell damage is still understood incompletely. Here, we demonstrated that propofol protects endothelial cells from inflammation and ROS damage via ABCA1-mediated cholesterol efflux after HDL incubation.
Materials and Methods
Chemicals and reagents
Propofol (2,6-diisopropylphenol), 2′,7′-dichlorofluorescin diacetate (DCF-DA), and cholesterol were purchased from Sigma-Aldrich (St. Louis, MO). HDL was provided by Intracel Corporation (Frederick, MD).
Cell lines and cell culture
(RAECs) were purchased from Cell Applications (Cell Applications, Inc., San Diego, CA, USA). Briefly, cells were cultured in T-25 flasks (Cell Applications, Inc., San Diego, CA, USA) with rat endothelial cell growth medium with growth supplements (Cell Applications, Inc., San Diego, CA, USA). The culture medium was replaced every other day. Once the cells reached 60-70% confluence, they were trypsinised and seeded in 6- or 24-well plastic plates for the following experiments.
Measurement of intracellular ROS production
Cells were washed with phosphate-buffered saline (PBS) and incubated with 10 μM DCF-DA (Sigma) at 37°C for 30 min in the dark. In the presence of ROS, DCF-DA is oxidised and becomes fluorescent. After incubation, the cells were trypsinised and washed with ice-cold PBS three times. ROS were quantified by flow cytometry (BD Biosciences, CA, USA) with 488 nm excitation and 585 nm emission filters.
Analysis of superoxide generation
The measurement of the superoxide generated was dependent on the reduction of ferricytochrome c by using SOD Assay Kit-WST (Dojindo Molecular Technologies, USA) according to the manufacturer's instructions. Briefly, after treatment of cells with propofol, the cells were mixed with WST working solution and incubated at 37°C for 20 min. After incubation, SOD were quantified by readingwith the absorbance at 450 nm using a microplate reader (Fusion™, Packard BioScience, Waltham, MA, USA).
Measurement of intracellular glutathione (GSH) levels
Intracellular GSH levels were assayed by fluorescent monochlorobimane (mBCl; Molecular Probes, Eugene, OR, USA) according to the manufacturer's instructions. Briefly, after treatment of cells with propofol, the cells were re-incubated in DMEM containing 100 μM mBCl at 37 °C for 30 min in the dark. Fluorimetric analysis was performed using a fluorescence plate reader with 400 nm excitation and 505 nm emission filters (Fusion™, Packard BioScience, Waltham, MA, USA).
Transient transfection of siRNA
Rat ABCA1 siRNA and rat scrambled siRNA (Dharmacon, Lafayette, CO) were used to knockdown ABCA1 expression in the RAECs. Twenty-four hours before transfection, 3 × 104 cells were seeded per 24-well plate. After transfection at 37°C for 5 h, the cells were added to 250 μL of DMEM containing 20% serum and 100 μg/mL cholesterol and grown.
Western blot analysis
Cells were collected, washed three times with PBS, and lysed with RIPA lysis buffer (Pierce, Rockford, IL), containing 1% of the Sigma protease cocktail, for 30 min at 4°C. The lysates were centrifuged at 10,000 ×g and 4°C to obtain solubilised cellular proteins. The supernatant protein concentration was measured by a bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL). Proteins were separated by SDS-PAGE in a 6%, 10%, or 12% gel and electrotransferred onto a polyvinylidene fluoride membrane. Blots were probed with specific primary antibodies, followed by a horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody (1:5000) or HRP-conjugated goat anti-mouse IgG antibody (1:5000) (Zymed, CA, USA). After a wash with PBS containing 0.5% of Tween 20, peroxidase activity was assessed using enhanced chemiluminescence (ECL; PerkinElmer Life Science, MA, USA). The same membrane was re-probed with a monoclonal antibody against β-actin as a loading control (1:5000; Santa Cruz, Dallas, TX, USA). The intensities of the immunoreactive bands were analysed in the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Nuclear fraction extraction
The cells were collected and resuspended in a hypotonic buffer (10 mM 4-[2-hydroxyethyl]-1-piperazineethanesulfonate [HEPES]-KOH, pH 7.9; 10 mM KCl; 1.5 mM MgCl2; 0.2 mM phenylmethylsulfonyl fluoride; 20 μg/mL aprotinin; 0.5 mM dithiothreitol; and 0.5% NP-40) on ice for 15 min. After centrifugation at 6,000 ×g for 15 min at 4°C, the pellet was collected and then washed with basal buffer (hypotonic buffer without 0.5% NP-40). After centrifugation again at 6,000 ×g for 15 min at 4°C, the pellet was collected and resuspended in hypertonic buffer (20 mM HEPES-KOH, pH 7.9; 400 mM KCl; 1.5 mM MgCl2; 0.2 mM phenylmethylsulfonyl fluoride; 20 μg/mL aprotinin; 0.5 mM dithiothreitol; 0.2 mM EDTA; 10% glycerol) at room temperature for 30 min. After centrifugation at 10,000 ×g for 30 min at 4°C, the nuclear fraction contained in the supernatant was collected.
Cholesterol-loaded cells
These cells were prepared as described previously [13, 14]. Briefly, subconfluent monolayers of endothelial cells were washed twice with PBS containing 2 mg/mL fatty acid-free albumin (FAFA; Sigma) and incubated with DMEM containing 2 mg/mL FAFA and 50 μg/mL cholesterol in ethanol (10 mg/mL) for 48 h at 37°C.
Cholesterol efflux
The cholesterol efflux assay was carried out as described elsewhere [13, 14], with minor modifications. The cells grown in 6-well plates were incubated with DMEM containing 2 mg/mL FAFA and 0.25 μCi/mL [3H]cholesterol for 24 h. Before the efflux experiment, the cells were washed with DMEM-FAFA and incubated with DMEM-FAFA containing HDL (50 μg/mL; Intracel) or BSA (2 mg/mL) at 37°C for 30 min. After that, the media were collected, and the cells were solubilised in 0.5N NaOH for 5 h at room temperature. The radioactivity of the media and cell extracts was measured using TOPcount machinery (Beckman, CA, USA). The results represent radioactivity in the media as a percentage of total radioactivity (media + cell lysate) [13-15].
Detection of NF-κB p65 activity
To determine the NF-κB p65 activity, we used NF-κB p65 Transcription Factor Assay Kit (ab133112, abcam, MA, USA) as an approach. According to the manufacturer's instructions, briefly, the nuclear protein extraction was collected as described earlier and used for the determination of intracellular p65-NF-κB activity by spectrophotometer reader with absorbance at OD 450 nm. All measured values were detected by Synergy HT (BioTek, VT, USA).
Detection of PPARγ activity
We detected the PPARγ activity by using the PPARγ Transcription Factor Assay Kit (ab133101, abcam, MA, USA) according to the manufacturer's protocol. Briefly, he nuclear protein extraction was collected as described earlier and used for the determination of PPARγ activity activity by spectrophotometer reader with absorbance at OD 450 nm. All measured values were detected by Synergy HT (BioTek, VT, USA).
Statistical analysis
Data are expressed as means ± SEM. Groups were compared by one-way or two-way ANOVA followed by Bonferroni's post hoc analysis. Data with p < 0.05 were considered statistically significant.
Results
Propofol promotes HDL-induced ABCA1 expression in cholesterol-loaded RAECs
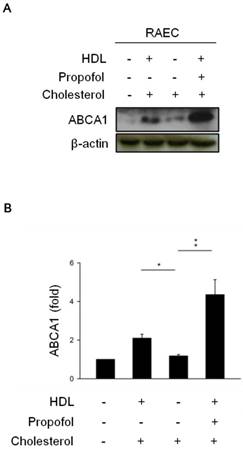
To verify the effect of propofol on the expression of ABCA1 in cholesterol-loaded RAECs after HDL incubation, we determined the expression of ABCA1 by immunoblotting in the differences among the groups, including control group (in absence of HDL-, cholesterol-, and propofol- loaded RACEs), cholesterol group (in presence of cholesterol- but in absence of HDL-and propofol loaded RACEs), HDL-cholesterol group (in presence of HDL- and cholesterol-, but in absence of propofol- loaded RACEs), and propofol group (in presence of HDL-, cholesterol- and 30 mmol/L propofol- loaded RACEs). The results showed that the expression of ABCA1 was more increased in HDL-cholesterol group (Lane 2, Figure 1) and dramatically increased in propofol group (Lane 4, Figure 1) than cholesterol group (Lane 3, Figure 1).
The effect of propofol on ABCA1 expression. (A) HDL induced ABCA1 expression in cholesterol-loaded RAECs, and propofol enhanced ABCA1 expression after HDL treatment. β-Actin served as a loading control. (B) Quantitative results on the level of ABCA1. All data are presented as the mean ± SEM, n = 3, *p < 0.05; **p < 0.01.
Propofol increases cholesterol efflux via ABCA1 induction
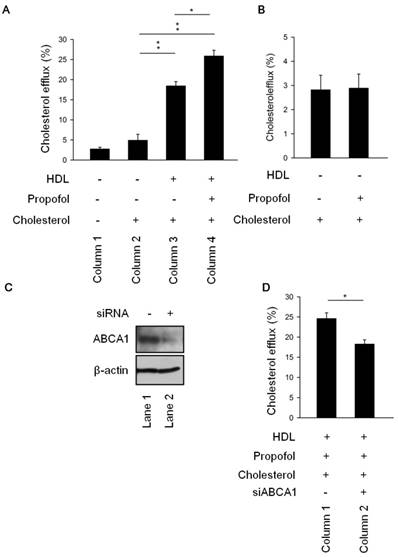
To further elucidate the role of propofol in cholesterol efflux, we measured the cholesterol efflux in the differences four groups. The results revealed that cholesterol efflux was more rapid in HDL-cholesterol group (Column 3, Figure 2A) than cholesterol group (Column 2, Figure 2A). Of note, as shown in Figure 2A (Column 4), treatment with propofol (propofol group) increased the cholesterol efflux than cholesterol group (Column 2, Figure 2A). Moreover, propofol could not promote cholesterol efflux in absence of HDL (Figure 2B). To further speculate whether the increasing in cholesterol efflux was caused by propofol through ABCA1 induction, we detected cholesterol efflux in knockdown ABCA1 (siABCA1) or not (scramble) RACEs (Figure 2C). Results demonstrated that ABCA1 knockdown decreased propofol induced cholesterol efflux (Figure 2D). We suggested that propofol increased cholesterol efflux via ABCA1 induction upon HDL loaded-RACEs.
Propofol enhanced cholesterol efflux via ABCA1 induction upon HDL-loaded RACEs. (A)After incubation with or without 30 mmol/L propofol for 1 h, RAECs were cultured in the presence of [3H]cholesterol (0.5 μCi/mL) for 24 h. Cholesterol efflux was initiated by incubating RAECs with HDL (50 μg/mL) for 30 min. The levels of cholesterol efflux were analysed. (B) Propofol alone could not promote cholesterol efflux without HDL-treatment. (C) Immunoblot analysis showing the expression levels of ABCA1 and caveolin-1 in ABCA1 siRNA and scrambled siRNA cells. (D) The levels of cholesterol efflux In ABCA1 siRNA cells and scrambled siRNA cells. All data are presented as the mean ± SEM, n = 3, *p < 0.05; **p < 0.01.
Propofol attenuates ROS production, superoxide generation, glutathione depletion, and activation of NF-kB pathway
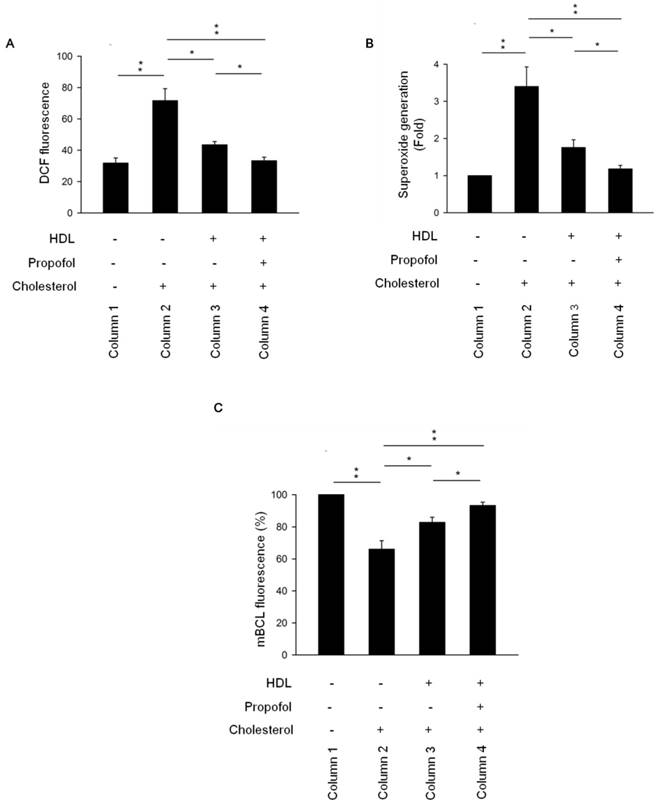
Galkina and Ley proposed that nitric oxide production by endothelial cells decreases and the burden of ROS increases under proatherogenic conditions [16]. Disruption of ROS-generating NADPH oxidase has beneficial effects against atherosclerosis. Additionally, ROS are key signalling molecules that play an important role in the progression of inflammatory disorders [17] and lead to impaired vascular function. Inhibitory targeting of inflammatory molecules and ROS also reduces atherosclerosis [18]. According to Figure 3, an increase in ROS production was observed in cholesterol group (Column 2, Figure 3A) but was decreased in HDL-cholesterol and propofol groups (Columns 3 and 4, Figure 3A). To further confirm the effect of propofol on ROS production, we detected the superoxide production and intracellular GSH levels. Results showed that superoxide generation was increased in cholesterol group (Column 2, Figure 3B), but was decreased in HDL-cholesterol and propofol groups (Columns 3 and 4, Figure 3B). Interestingly, propofol has a highest inhibitory effect on superoxide generation (Column 4, Figure 3B). Moreover, GSH concentration has an important effect on intracellular redox homeostasis. To investigate whether propofol prevent intracellular GSH depletion in cholesterol- loaded cells, the amount of intracellular GSH was assayed. Results showed that GSH level was decreased in cholesterol group (Column 2, Figure 3C), but was increased in HDL-cholesterol and propofol groups (Columns 3 and 4, Figure 3C). Propofol has highest protective effect on GSH depletion (Column 4, Figure 3C).
Propofol enhanced HDL-alleviated ROS production in cholesterol-loaded RAECs. (A) The level of intracellular ROS was determined by the DCF-DA assay, and the fluorescence was detected by FACS Calibur analysis. ROS formation was determined via mean fluorescence intensity. (B) Superoxide generation was determined by SOD Assay Kit-WST and measured by spectrophotometry at 450 nm. (C) Intracellular GSH levels were determined using mBCl and using a fluorescence plate reader with 400 nm excitation and 505 nm emission filters. All data are presented as mean ± SEM, n = 3, *p < 0.05; **p < 0.01.
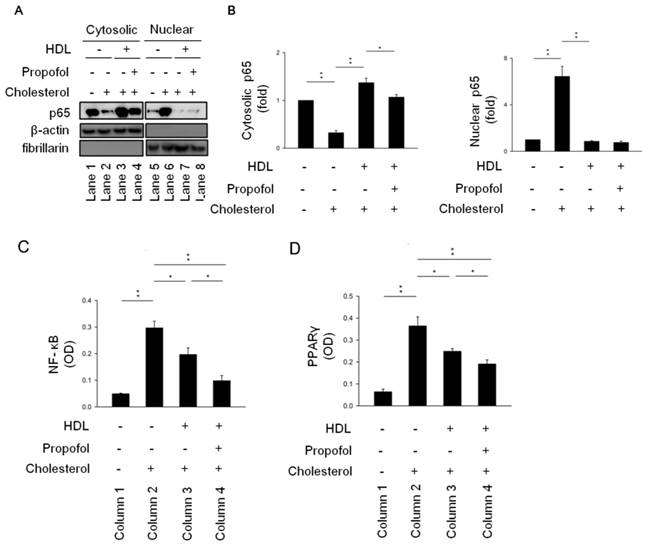
Fountain et al. demonstrated that NF-κB-p65 is detectable by western blotting of cytoplasmic and nuclear-protein fractions [19]. Our results showed that the level of NF-κB activation significantly increased in cholesterol group (Line 2, Figure 4A), as revealed by the increase in nuclear localisation of NF-κB subunit p65 with a concomitant decrease in cytosolic localisation in cholesterol group (Lines 2 and 6, Figure 4A). Furthermore, nuclear localisation of NF-κB in cholesterol-loaded RAECs after HDL incubation in the absence (HDL-cholesterol group, Line 7, Figure 4A) or presence of propofol treatment (propofol group, Line 8, Figure 4A) decreased to the background level (Figure 4A). However, there are no difference between HDL-cholesterol group and propofol group (Figure 4B). To further demonstrated the role of propofol on NF-κB activity, we detected its activity as described earlier. Results showed that propofol has highest inhibitory effect on NF-κB activity (Column 4, Figure 4C). Consequently, we suggested that propofol attenuates ROS production and NF-κB activation.
Propofol alleviated cholesterol-caused activation of NF-kB pathway. (A) RAECs were collected, and cytosolic and nuclear fractions were isolated as described in Methods. A western blot was carried out to detect the subcellular localisation of NF-κB with an antibody against the NF-κB subunit p65. β-Actin served as a cytosolic marker, and fibrillarin as a nuclear marker. NF-κB nuclear localisation was higher both in HDL- and propofol-treated RAECs after cholesterol incubation. (B) Quantitative results on the level of cytosolic and nuclear NF-κB. Relative (C) NF-κB-luciferase and (D) PPARγ activities were detected in the indicated four groups. All data are presented as the mean ± SEM, n = 3, *p < 0.05; **p < 0.01.
Discussion
Atherosclerosis is an inflammatory disease of the wall of large- and medium-sized arteries that is precipitated by elevated levels of LDL cholesterol in blood [16, 20]. ABCA1 plays an important role in artery wall cell-mediated modification/oxidation of LDL by modulating the release of ROS from artery wall cells; these compounds are necessary for LDL oxidation [21]. Moreover, caveolae and caveolin-1 are on the centre stage of cholesterol transport and inflammation in macrophages [22]. Cholesterol efflux is closely related to the expression levels of Cav-1 and ABCA1 [22, 23]. Chao et al. provided evidence for a direct interaction between ABCA1 and HDL, ABCA1 and caveolin-1, but not HDL and caveolin-1, indicating that ABCA1 may act as a structural platform for the interaction between HDL and caveolin-1 on the cell surface during cellular cholesterol efflux [24]. Pavlides et al. provided clear evidence that the absence of Cav-1 in macrophages is pro-atherogenic, whereas its absence in endothelial cells protects against formation of atherosclerotic lesions [25]. Lin et al. have reported that the interaction between caveolin-1 and ABCA1 performs an important function in the transport of lipids between the Golgi apparatus and plasma membrane caveolae [13].
Ma et al. suggested that anti-inflammatory cytokine IL-10 was decreased and pro-inflammatory cytokines (TNFα, IL-6, IL-12, and GCSF) was increased in ABCA1-/- mouse. Furthermore, cholesterol enrichment decreased CREB phosphorylation and promote pro-inflammatory response. Disrupting lipid rafts by statins, methyl-β-cyclodextrin or filipin also activates PKA signaling pathway and recuperated ABCA1 phenotype and likely functions downstream of ABCA1 [26]. p65 is well known as an indicator of activation of the NF-κB pathway and proposed to be a substrate of PKA [27, 28]. Therefore, we proposed that propofol up-regulated ABCA1 and attenuated inflammation via PKA-p65. However, we need more evidences to prove it.
LXR-RXR heterodimers have anti-inflammatory effects by upregulating ABCA1, ABCG1, and SR-B1 promoting the efflux of cholesterol from macrophages and thus may counter the amplification of TLR signalling by cellular cholesterol accumulation after propofol treatment [7, 20]. However, there is no evidence to study the effect of propofol on PPARγ/LXRα pathway in RACEs. Therefore, we detected the PPARγ activity by PPAR gamma Transcription Factor Assay Kit (ab133101, abcam) (Figure 4D). Results showed that propofol has highest inhibitory effect on PPARγ activity (Column 4, Figure 4D). Additionally, propofol has been used as an antioxidant in a porcine ischemia-reperfusion model [29]. In a clinical study, small-dose propofol sedation attenuated free-radical production after a release of the tourniquet during total knee replacement under spinal anaesthesia [30]. In the present study, our results showed that propofol reduced ROS production in cholesterol-loaded RAECs (Figure 3) in line with findings in other studies [31-33]. Furthermore, we demonstrated that propofol promoted cholesterol efflux after HDL treatment (Figure 2); however, Adaramoye et al. demonstrated that this treatment elicited a detrimental effect on the lipid profile resulting in hypercholesterolaemia, which subsequently leads to abnormally high activities of serum creatinine phosphokinase and lactate dehydrogenase in rats [34]. On the other hand, it has been reported that ABCA1, ABCG1, and SR-B1 was up-regulated by propofol via the PPAR-γ/LXRα signalling pathway in THP-1 macrophage-derived foam cells [7]. Therefore, the critical influence of propofol on cholesterol efflux is still controversial. Future studies should further delineate the exact effects of propofol on this event.
Finally, our data point to the protective role of propofol against endothelial-cell inflammation and ROS damage via upregulation of ABCA1-mediated cholesterol efflux after HDL incubation.
Acknowledgements
This study was supported by grant RD106059 from Show Chwan and Chan Bing Show Chwan Memorial Hospital, Changhua, Taiwan.
Author Contributions
C.P.H., C.H.L., and C.Y.K. designed and performed the experiments, derived the models and analyzed the data. C.Y.K. wrote and revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hoeke G, Kooijman S, Boon MR, Rensen PC, Berbee JF. Role of Brown Fat in Lipoprotein Metabolism and Atherosclerosis. Circ Res. 2016;118:173-82
2. Wang HH, Garruti G, Liu M, Portincasa P, Wang DQ. Cholesterol and Lipoprotein Metabolism and Atherosclerosis: Recent Advances In reverse Cholesterol Transport. Ann Hepatol. 2017;16:21-36
3. Yu XH, Fu YC, Zhang DW, Yin K, Tang CK. Foam cells in atherosclerosis. Clin Chim Acta. 2013;424:245-52
4. Karr S. Epidemiology and management of hyperlipidemia. Am J Manag Care. 2017;23:S139-S48
5. Marik PE. Propofol: an immunomodulating agent. Pharmacotherapy. 2005;25:28S-33S
6. Allaouchiche B, Debon R, Goudable J, Chassard D, Duflo F. Oxidative stress status during exposure to propofol, sevoflurane and desflurane. Anesth Analg. 2001;93:981-5
7. Ma X, Li SF, Qin ZS, Ye J, Zhao ZL, Fang HH. et al. Propofol up-regulates expression of ABCA1, ABCG1, and SR-B1 through the PPARgamma/LXRalpha signaling pathway in THP-1 macrophage-derived foam cells. Cardiovasc Pathol. 2015;24:230-5
8. Lin C, Sui H, Gu J, Yang X, Deng L, Li W. et al. Effect and mechanism of propofol on myocardial ischemia reperfusion injury in type 2 diabetic rats. Microvasc Res. 2013;90:162-8
9. Wang S, Smith JD. ABCA1 and nascent HDL biogenesis. Biofactors. 2014;40:547-54
10. Rosenson RS, Brewer HB Jr, Ansell BJ, Barter P, Chapman MJ, Heinecke JW. et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol. 2016;13:48-60
11. Ooi BK, Goh BH, Yap WH. Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. Int J Mol Sci. 2017:18
12. Catapano AL, Pirillo A, Norata GD. Vascular inflammation and low-density lipoproteins: is cholesterol the link? A lesson from the clinical trials. Br J Pharmacol. 2017;174:3973-85
13. Lin YC, Ma C, Hsu WC, Lo HF, Yang VC. Molecular interaction between caveolin-1 and ABCA1 on high-density lipoprotein-mediated cholesterol efflux in aortic endothelial cells. Cardiovasc Res. 2007;75:575-83
14. Kuo CY, Lin YC, Yang JJ, Yang VC. Interaction abolishment between mutant caveolin-1(Delta62-100) and ABCA1 reduces HDL-mediated cellular cholesterol efflux. Biochem Biophys Res Commun. 2011;414:337-43
15. O'Connell BJ, Denis M, Genest J. Cellular physiology of cholesterol efflux in vascular endothelial cells. Circulation. 2004;110:2881-8
16. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. 2009;27:165-97
17. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126-67
18. Wadley AJ, Veldhuijzen van Zanten JJ, Aldred S. The interactions of oxidative stress and inflammation with vascular dysfunction in ageing: the vascular health triad. Age (Dordr). 2013;35:705-18
19. Fountain MD, Abernathy LM, Cannon AC, Joiner MC, Hillman GG. Inhibition of radiation-induced lung inflammation and NF-kB activation by soy isoflavones. J Immunol. 2017:198
20. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104-16
21. Reddy ST, Hama S, Ng C, Grijalva V, Navab M, Fogelman AM. ATP-binding cassette transporter 1 participates in LDL oxidation by artery wall cells. Arterioscler Thromb Vasc Biol. 2002;22:1877-83
22. Qin L, Zhu N, Ao BX, Liu C, Shi YN, Du K. et al. Caveolae and Caveolin-1 Integrate Reverse Cholesterol Transport and Inflammation in Atherosclerosis. Int J Mol Sci. 2016;17:429
23. Luscher TF, Landmesser U, von Eckardstein A, Fogelman AM. High-density lipoprotein: vascular protective effects, dysfunction, and potential as therapeutic target. Circ Res. 2014;114:171-82
24. Chao WT, Tsai SH, Lin YC, Lin WW, Yang VC. Cellular localization and interaction of ABCA1 and caveolin-1 in aortic endothelial cells after HDL incubation. Biochem Biophys Res Commun. 2005;332:743-9
25. Pavlides S, Gutierrez-Pajares JL, Katiyar S, Jasmin JF, Mercier I, Walters R. et al. Caveolin-1 regulates the anti-atherogenic properties of macrophages. Cell Tissue Res. 2014;358:821-31
26. Ma L, Dong F, Zaid M, Kumar A, Zha X. ABCA1 protein enhances Toll-like receptor 4 (TLR4)-stimulated interleukin-10 (IL-10) secretion through protein kinase A (PKA) activation. J Biol Chem. 2012;287:40502-12
27. Wall EA, Zavzavadjian JR, Chang MS, Randhawa B, Zhu X, Hsueh RC. et al. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci Signal. 2009;2:ra28
28. Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413-24
29. Hsiao HT, Wu H, Huang PC, Tsai YC, Liu YC. The effect of propofol and sevoflurane on antioxidants and proinflammatory cytokines in a porcine ischemia-reperfusion model. Acta Anaesthesiol Taiwan. 2016;54:6-10
30. Cheng YJ, Wang YP, Chien CT, Chen CF. Small-dose propofol sedation attenuates the formation of reactive oxygen species in tourniquet-induced ischemia-reperfusion injury under spinal anesthesia. Anesth Analg. 2002;94:1617-20 table of contents
31. Meng T, Yu J, Lei Z, Wu J, Wang S, Bo Q. et al. Propofol reduces lipopolysaccharide-induced, NADPH oxidase (NOX 2) mediated TNF- alpha and IL-6 production in macrophages. Clin Dev Immunol. 2013;2013:325481
32. Yang SC, Chung PJ, Ho CM, Kuo CY, Hung MF, Huang YT. et al. Propofol inhibits superoxide production, elastase release, and chemotaxis in formyl peptide-activated human neutrophils by blocking formyl peptide receptor 1. J Immunol. 2013;190:6511-9
33. Hsing CH, Lin MC, Choi PC, Huang WC, Kai JI, Tsai CC. et al. Anesthetic propofol reduces endotoxic inflammation by inhibiting reactive oxygen species-regulated Akt/IKKbeta/NF-kappaB signaling. PLoS One. 2011;6:e17598
34. Adaramoye OA, Akinwonmi O, Akanni O. Effects of propofol, a sedative-hypnotic drug, on the lipid profile, antioxidant indices, and cardiovascular marker enzymes in wistar rats. ISRN Pharmacol. 2013;2013:230261
Author contact
![]() Corresponding author: Chan-Yen Kuo, Graduate Institute of Systems Biology and Bioinformatics, National Central University, Chung-li, Taiwan. Department of Ophthalmology, Hsin Sheng Junior College of Medical Care and Management, Longtan, Taiwan. E-mail address: cykuoedu.tw. Tel.: +886 3-4227151; ext: 36115. Fax: +886 3-4226062.
Corresponding author: Chan-Yen Kuo, Graduate Institute of Systems Biology and Bioinformatics, National Central University, Chung-li, Taiwan. Department of Ophthalmology, Hsin Sheng Junior College of Medical Care and Management, Longtan, Taiwan. E-mail address: cykuoedu.tw. Tel.: +886 3-4227151; ext: 36115. Fax: +886 3-4226062.