Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(8):771-781. doi:10.7150/ijms.24635 This issue Cite
Research Paper
The replicative senescent mesenchymal stem / stromal cells defect in DNA damage response and anti-oxidative capacity
Jin Yu1, Jiazhong Shi1, Yue Zhang1,2, Yi Zhang1, Yaqin Huang1, Zhiwen Chen3, Jin Yang1 ![]()
1. Department of Cell Biology, Army Medical University (The Third Military Medical University), Chongqing 400038, China
2. Department of Pathology, The 451th hospital of PLA, Xi'an 710000, China
3. Urology Institute of PLA, Southwest Hospital, Army Medical University (The Third Military Medical University), Chongqing 400038, China
Received 2017-12-28; Accepted 2018-4-27; Published 2018-5-22
Abstract
Replicative senescence and potential malignant transformation are great limitations in the clinical application of bone marrow-derived mesenchymal stem / stromal cells (MSCs). An abnormal DNA damage response may result in genomic instability, which is an integral component of aging and tumorigenesis. However, the effect of aging on the DNA damage response in MSCs is currently unknown. In the present study, we evaluated the DNA damage response induced by oxidative stress and DNA double-strand breaks in human bone marrow-derived MSCs. After long-term cell culture, replicative senescent MSCs (sMSCs) were characterized by a poor proliferation rate, high senescence-associated β-galactosidase activity, and enhanced expression of P53 and P16. Features of the DNA damage response in these sMSCs were then compared with those from early-passage MSCs. The sMSCs were more sensitive to hydrogen peroxide and bleomycin treatment with respect to cell viability and apoptosis induction. Combined with the comet assay, γH2AX foci characterization and reactive oxygen species detection were used to demonstrate that the antioxidant and DNA repair ability of sMSCs are attenuated. This result could be explained, at least in part, by the downregulation of anti-oxidation and DNA repair genes, including Cu/Zn-SOD, GPX, CAT, OGG1, XRCC1, Ku70, BRCA2 and XRCC4. In conclusion, MSCs aging is associated with a reduction in the DNA repair and anti-oxidative capacity.
Keywords: mesenchymal stem cells, DNA damage response, Replicative senescence, oxidative stress, DNA double-strand breaks
Introduction
Mesenchymal stem /stromal cells (MSCs) are a multipotent adult stem cell population that, given appropriate culture conditions, are able to self-replicate and differentiate into a variety of cell types such as osteoblasts, chondrocytes, adipocytes, and smooth muscle cells [1, 2]. Although isolated and cultured MSCs according to International Society for Cellular Therapy (ISCT) criteria are heterogeneous and contain several populations, they are currently served as sources of putative MSCs for therapeutic purpose[3]. Human MSCs have gained attention because of their therapeutic potential in the context of regenerative medicine [4]. A major source of human MSCs is adult bone marrow. Due to their limited number, MSCs obtained from bone marrow require expansion in vitro before being transplanted for tissue regeneration. However, similar to any somatic cell, the MSC has a limited lifespan in vitro and becomes senescent after a certain number of cell divisions, which is associated with a deterioration of the regenerative potential [5]. Moreover, previous studies have indicated that extensive culture of various MSCs of animal origin can cause spontaneous transformation [6-8]. Long-term culture has also been suspected of inducing oncogenic transformation of human MSCs [9]. Therefore, the safety of the therapeutically promising human MSC should be carefully defined before the cells are used in the clinical setting, and the study of the potential transformation of human MSCs and the associated molecular mechanism is of great value for clinical application.
Although the mechanism of the transition from a senescent to a malignant cell is unknown, previous studies have shown that embryonic stem cells, MSCs and induced pluripotent stem cells expanded in vitro exhibit genomic instability[7, 10, 11]. Genomic instability in turn confers an increased risk of malignant transformation that may negatively affect the safety of cultured stem cell transplantation. The accumulation of DNA damage has been implicated as an important mechanism governing the aging process as determined by in vitro studies of human cells and animal models containing engineered defects in diverse DNA repair pathways [12-14]. Knock-down of DNA repair genes, such as FEN1, RAD51, EXO1, BRCA1 alone was able to induce premature senescence in human fibroblasts, silencing of RB1 induces cellular senescence and impairs the differentiation potential of human MSCs[15-17]. Additionally, defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells, in vitro senescence of rat MSCs is accompanied by the down regulation of stemness-related and DNA damage repair genes [18, 19]. Thus, the involvement of an abnormal DNA damage response in the generation of senescent human MSCs (sMSCs) requires analysis. Such studies may provide insight to help clarify the mechanisms of genomic instability and malignant transformation in these cells.
To determine the specific DNA damage response properties of sMSCs after extensive culture, we compared sMSCs with early-passage MSCs (young MSCs, yMSCs) with respect to the DNA damage response arising from oxidative stress and DNA double-strand breaks. Our results show that the replicative senescence of MSCs is accompanied by a defective, decreased antioxidant ability and DNA damage response.
Materials and Methods
hMSC isolation and cell culture
Human bone marrow stem cells /stromal cells (BMSCs) were collected from 3 healthy volunteers. All procedures were approved by the Ethics Committee at Third Military Medical University. BMSCs were obtained and processed as described previously [20]. Briefly, BMSCs were cultured in alpha-MEM (Hyclone, USA) supplemented with antibiotics and 10% fetal bovine serum. Cultures were passaged when they reached 75% to 80% confluence. The initial confluent culture was designated 'passage 0' (P0). From the 1st passage onward, the level of population doubling (PD) and the population doubling time (PDT) were calculated based on the total cell number and the time between passages.
Immunophenotyping of cultured MSCs
MSCs were incubated with anti-CD19, anti-CD146, anti-CD44, anti-CD45, anti-CD90, and anti-CD105 antibodies (R&D, USA) at room temperature for 30 min. After washing twice with PBS, the MSCs were incubated with a FITC-labeled secondary antibody in the dark for 30 min. After washing, the cells were suspended in PBS and analyzed on a flow cytometer.
Differentiation assays
For osteogenesis, the cultures were incubated in osteogenic differentiation medium (R&D, USA). The medium was replaced two times per week for 2 weeks. The cells were fixed with 2% formalin for 20 min at room temperature (RT) and stained with Alizarin Red, pH 4.1 (Sigma, USA) for 20 min at RT. For adipogenesis, the cultures were incubated in adipogenic differentiation medium (R&D, USA). The medium was changed two times per week for 2 weeks. The cells were fixed with 4% formalin for 10 min at RT and stained with 0.5% Oil Red O (Sigma, USA) in methanol for 20 min at RT.
Senescence-associated β-galactosidase staining
Senescence-associated β-galactosidase (SA-β-gal) activity was determined using an SA-β-gal Staining Kit from Beyotime Technology (China) according to the manufacturer's instructions. Briefly, cells were fixed for 15 min at room temperature and then incubated in staining solution at 37°C overnight. Senescent cells were identified as blue-stained cells by standard light microscopy.
Cytotoxicity testing
Cytotoxicity was measured using the CCK-8 kit (Roche, Germany) according to the manufacturer's instructions. Briefly, cells were seeded into flat-bottom 96-well plates at the density of 3×103 cells per well. After 24 hours, cells were treated with the DNA-damaging agents H2O2 for 4 hours or BLM for 24 hours. The cells were cultured for another 2-3 days for the CCK-8 assay. The colorimetric readout values were normalized relative to untreated controls. The data shown were obtained from three independent experiments.
ROS detection
The level of intracellular reactive oxygen species (ROS) was measured using DCFH-DA (Beyotime Technology, China). DCFH-DA is cleaved intracellularly by nonspecific esterases to form DCFH, which is further oxidized by ROS to form the fluorescent compound DCF. The DCFH-DA working solution was added directly to the medium for a final concentration of 10 μM, and the cells were then incubated at 37°C for 15 min. Images were captured using a Nikon camera (Nikon, Japan) under 200× magnification from 10 randomly selected fields containing 200-300 cells.
Real-time PCR
The total RNA of MSCs was extracted using the RNeasy Mini Kit (QIAGEN, Germany). Reverse transcription of 1 μg of RNA was completed using the High-Capacity cDNA RT Kit (Bio-Rad, USA) following the manufacturer's instructions. Real-time quantitative PCR was performed on the CFX Connect Real-Time system (Bio-Rad, USA) using the SYBR Green Supermix (Bio-Rad, USA). All runs were completed in triplicate, and the results are presented as the average of three separate runs, with quantification performed by normalizing to GAPDH expression.
Comet assay (alkaline condition)
Cells were resuspended in PBS (Ca2+- and Mg2+-free) at a concentration of 1×105 cells/ml. Briefly, an aliquot of 50 μl of cells (1×105 cells/ml) was added to 500 μl of molten LMA agarose (1% low-melting agarose) maintained at 42°C. The mixture was immediately pipetted and evenly distributed onto a comet slide. The slide was incubated at 4°C in the dark for 1 min to accelerate gelling of the agarose disc and then transferred into pre-chilled alkaline lysis solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris-base, 10% DMSO, 1% Triton X-100, pH 10) for 2 hours at 4°C. A denaturation step was performed in alkaline solution (300 mM NaOH, 1 mM EDTA, pH>13) at room temperature for 20 min in the dark. The slide was then transferred to pre-chilled alkaline electrophoresis solution pH>13 (300 mM NaOH, 1 mM EDTA) and subjected to electrophoresis at 1 V/cm, 300 mA for 40 min in the dark at 4°C. Following electrophoresis, the slide was washed with neutralization buffer (0.4 M Tris-HCl, pH 7.4), immersed in ice cold 100% ethanol at room temperature for 5 min and air-dried. DNA was stained with ethidium bromide solution (Sigma, USA) for 20 min in the refrigerator and immediately analyzed using a Nikon fluorescence microscope. More than 100 comets were scored per sample using the CASP software.
Comet assay (neutral condition)
The procedure is essentially same as described in alkaline condition with slight modifications. Specifically, the gelled slide was incubated with prechilled neutral lysis solution (2.5 M NaCl, 30 mM Na2EDTA, 10 mM Tris-base, 1% DMSO, 1% Triton X-100, pH 7.5) for 2 hours at 4°C. After lysis, the slides were incubated in TBE for 20 min and electrophoresed at 25 V (1.0 V/cm) for 20 min. Then, the slides were washed in deionized water for 5 min and immersed in ice cold 100% ethanol for 5 min. Finally, the DNA was stained with ethidium bromide solution (Sigma, USA) and analyzed using Nikon fluorescence microscope. More than 100 comets were scored per sample using the CASP software.
Immunofluorescence and quantification of γH2AX foci
MSCs were cultured on coverslips and treated with a dose of 30 μg/ml BLM for 2 h. The coverslips were fixed with 3% paraformaldehyde (15 min, RT), permeabilized by incubation with 0.1% Triton in PBS (15 min, RT), blocked with 1% bovine serum albumin (30 min, RT), and incubated with a mouse anti-γH2AX monoclonal antibody (Abcam, UK) (overnight, 4°C). Primary antibodies were visualized by incubation with appropriate FITC-labeled secondary antibodies (Invitrogen, Germany), and the nuclei were counterstained with DAPI (Invitrogen, Germany). Images were obtained using a Nikon microscope, and the number of foci was counted using ImageJ software. To quantify γH2AX foci formation, 100-200 nuclei were evaluated for each simple.
Detection of apoptosis by fluorescence microscopy
MSCs were cultured on coverslips and treated with either 30 μg/ml BLM for 24 hours or 600 μM H2O2 for 4 hours. At 24 hours after H2O2 or BLM treatment initiation, MSCs were washed once with pre-chilled PBS and incubated with Annexin V Reagent (Beyotime Technology, China) in the dark for 10 min at RT. Then, the cells were washed once with 1× Binding Buffer (Beyotime Technology, China) and incubated with propidium iodide for 5 min at RT. Cells were observed using a Nikon fluorescence microscope. A total of 500 cells were evaluated for quantification of cell apoptosis for each data point.
Statistical analyses
All experiments were performed at least three times. The data were analyzed using Student's t-test. Significance was accepted at P<0.05.
Results
Characterization of human MSCs during long term in vitro culture
MSCs were isolated and cultured as described [20]. Cells with a typical fibroblast-like morphological homogeneity appeared after 10 PD. When the PD reached about 30, the cells exhibited a pronounced increase in size and had enlarged nuclei, all of which are characteristics of senescent cells (Fig. 1A). The growth kinetics of the MSC culture was examined and presented by plotting PD relative to culture time. Cell proliferation was slow during the early stage of culture as the PDT was 90.2±18.4 hours in the initial culture. PDT then gradually decreased and was maintained at a steady level of 41.2±2.6 hours. However, when the PD reached 35 to 40, the PDT gradually increased which was another characteristic of senescent cells (Fig. 1B).
Establishment of a replicative senescent MSCs model. A. morphology of human bone marrow derived MSCs during long term in vitro culture. B. Growth curve of cultured MSCs. C. FCM characterization of MSCs. D. Osteogenic and Adipogenic differentiation of MSCs was confirmed by staining with Alizarin red and Oil Red O, respectively. E. SA-β-gal staining of yMSCs, counter stained with hematoxylin. F. Real-time RT-PCR showing the expression of p16 and p53 in yMSCs and sMSCs. * indicated that p < 0.05, ** indicated that p < 0.01.
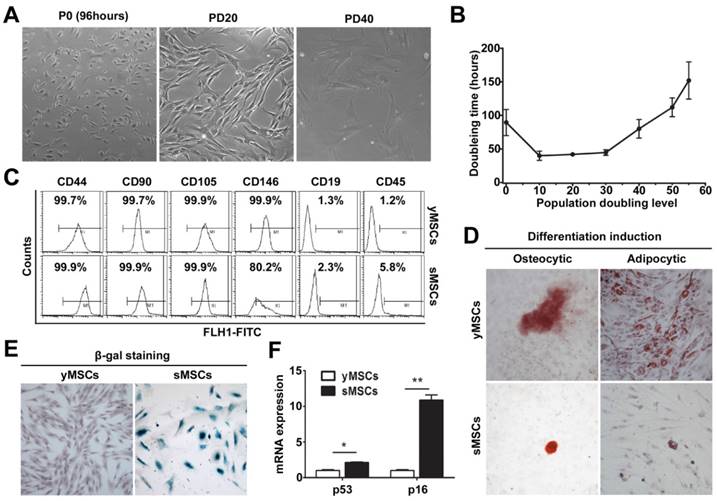
Cell surface markers were characterized by flow cytometry (FCM) at around 20 PD and 40 PD, which showed high expression of CD44, CD90, CD105 and CD146 and low expression of CD19 and CD45 (Fig.1C). Furthermore, adipocytic and osteocytic lineage induction was used to confirm the cells' capacity to differentiate (Fig. 1D). The senescence phenotype was assessed by SA-β-Gal activity (Fig. 1E) and was further confirmed by the expression of senescence-related genes P53 and P16 (Fig. 1F), all of which were strongly increased in MSCs at 40 PD compared with MSCs at 20 PD.
Taking together, these data indicate that MSCs were isolated, and a replicative senescence model was established by extensive in vitro culture. The MSCs at approximately 20 PD and 40 PD were chosen to represent yMSCs or sMSCs, respectively, in subsequent experiments.
The sMSCs were more sensitive to oxidative stress and DNA double-strand breaks
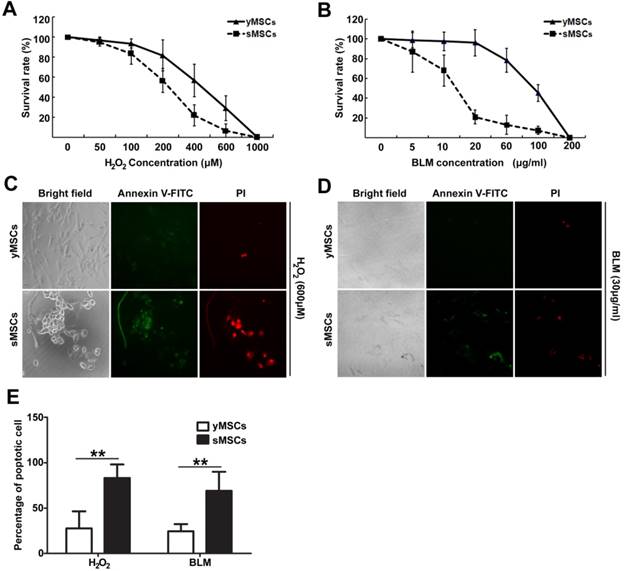
Endogeneous oxidative stress has been reported to induce cellular aging, which is manifested in decreased organ function [21, 22]. Meanwhile, MSCs often have DNA double-strand breaks caused by irradiation and chemical drugs during bone marrow implantation or anti-cancer treatment. To characterize the DNA damage response in senescent MSCs, we evaluated the biological outcomes of treatment of MSCs with two different DNA-damaging agents, hydrogen peroxide (H2O2) and bleomycin (BLM), which each induce DNA damage through oxidative stress and DNA double-strand breaks respectively.
Both H2O2 and BLM treatment caused markedly higher cytotoxicity in sMSCs (IC50 of 200 μM and 13 μg/ml, respectively) than in yMSCs (IC50 of 400 μM and 90 μg/ml, respectively) (Fig. 2A, B). To determine whether the decreased cell viability was due to induced apoptosis, apoptotic events were measured after exposure to H2O2 for 24 hours or BLM for 48 hours by staining Annexin V. As illustrated in Fig. 2 C, D and E, 27.6% and 24.4% of yMSCs were apoptotic after H2O2 and BLM treatment, respectively, compared with 83.2% and 69% of sMSCs. Therefore, compared to yMSCs, sMSCs are more sensitive to both H2O2 and BLM treatment.
sMSCs are more sensitive to H2O2 and BLM as compared with yMSCs. A and B. CCK-8 analysis of yMSCs and sMSCs after treatment of increasing concentration of H2O2 for 4h and cultured for 72h, or increasing concentration of BLM for 24h and cultured for 48h. C and D. Apoptosis detection in yMSCs and sMSCs. The cells were treated with 600μM of H2O2 for 4h and cultured for 24h (C) or treated with 30μg/ml BLM for 24h and cultured for 24h (D), and were then stained with Annexin V (green) and P I(red). E. Quantification of C and D. Data are presented as mean value±SD of three independent experiments. ** indicated that p < 0.01.
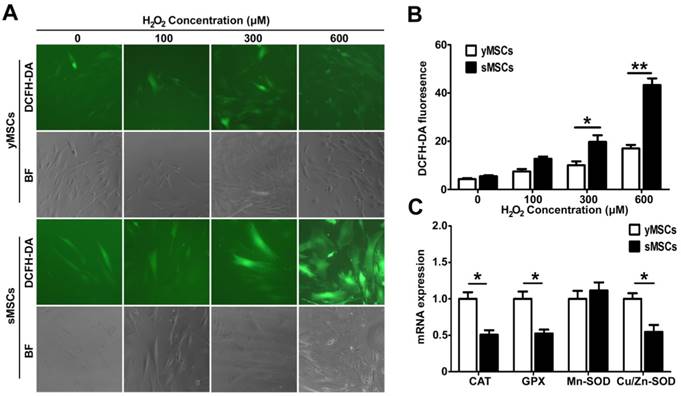
The antioxidant ability is decreased in sMSCs
Oxidative stress occurs largely due to ROS generated by exogenous factors or by cellular metabolism. ROS can interact with, or attack, multiple macromolecules including lipids, nucleic acids, and proteins. Several mechanisms are responsible for the protection of cells from potential cytotoxic damage caused by ROS. To determine MSC antioxidant ability, intracellular ROS levels in MSCs were measured after a 2 hours treatment with different doses of H2O2. As shown in Fig. 3 A and B, The DCF green fluorescence intensity increased significantly in a dose dependent manner after H2O2 treatment in both yMSCs and sMSCs. However, the DCF green fluorescence intensity in yMSCs are significantly higher than that in sMSCs after both 300 and 600 μM of H2O2 treatment (p<0.05 and P<0.01, respectively). This discrepancy in fluorescence indicates that the H2O2-induced ROS levels in sMSCs were substantially higher than that in yMSCs, which may be due to an attenuated antioxidant ability of sMSCs. Therefore, we analyzed the expression of major antioxidant genes, including Mn-SOD, Cu/Zn-SOD, GPX, and CAT, using real-time PCR (Fig. 3C). Consistent with the results of ROS production following H2O2 treatment, compared with yMSCs, there was a two-fold decrease in the expression levels of Cu/Zn-SOD, GPX, and CAT in sMSCs (P<0.05). However, no difference was observed in the expression of Mn-SOD between sMSCs and yMSCs.
The antioxidant ability is decreased in the sMSCs. A. The intracellular level of ROS was measured by the fluorescent dye DCFH-DA in MSCs without treatment(control), after 2h treatment with 100 μM, 300 μM or 600μM H2O2. B. The quantitation result of A. C. Real time-PCR analysis of the basal expression level of anti-oxdative Enzymes genes Cu/Zn-SOD、Mn-SOD, GPX and CAT in yMSCs and sMSCs. Data are presented as mean values±SD of three independent experiments. * indicated that p < 0.05, ** indicated that p < 0.01.
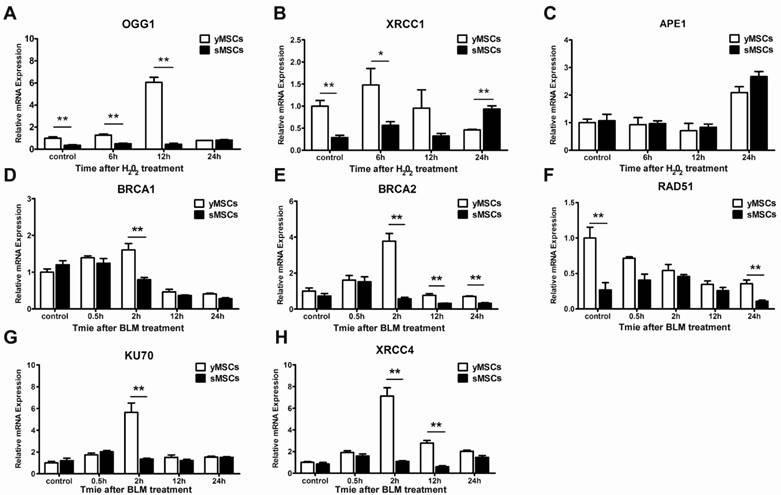
The expression of DNA repair genes is downregulated in sMSCs
Oxidative stress and DNA double-strand breaks can cause severe DNA damage, which are repaired by different DNA repair pathways. While oxidative stress induced DNA damages are mainly repaired by base excision repair (BER) [23], DNA double-strand breaks are mostly repaired by homologous recombinant repair (HR) or non-homologous end joining (NHEJ) [24]. To evaluate the DNA repair ability of MSCs, the expression levels of oxidative stress or double-strand-break-associated DNA repair genes, including OGG1, XRCC1 and APE1 for BER and Rad51, BRCA2, XRCC4, BRCA1, Ku70 for HR and NHEJ, were evaluated (Fig. 4A-H). Untreated cells were used to determine basal expression levels, and the expression of four genes were higher in yMSCs than in sMSCs: OGG1, XRCC1, RAD51, and BRCA2 (P<0.01) (Fig. 4 A, B, D, E). Moreover, after exposure to H2O2, OGG1 and XRCC1 were significantly expressed at a higher level in yMSCs than in sMSCs. Notably, sMSCs had almost no induction of OGG1 expression and a delayed induction of XRCC1 expression (Fig. 4F-H). On the other hand, after treatment with BLM, while Rad51 expression was downregulated both in yMSCs and sMSCs, the expression of BRCA1, Ku70, and XRCC4 genes was significantly induced only in yMSCs. Furthermore, the induced expression of BRCA2 declined more rapidly in sMSCs (Fig. 4 D-H). Collectively, the expression level of DNA repair genes was altered in sMSCs, which may contribute to a defective DNA damage response.
The expressions of DNA repair genes are downregulated in sMSCs. A to C. Real time-PCR analysis of BER genes including OGG1, XRCC1 and APE1 in cells at 6, 12 and 24 hours after 300 μM H2O2 treated for 2 hours. D, E. Real time-PCR analysis of DSB repair genes including BRCA1, BRCA2, Rad51, Ku70 and XRCC4 in cells at 0.5, 2, 6, 12 and 24 hours after 50μg/ml BLM treated for 2 hours. Data are presented as mean value±SD of three independent experiments. * indicated that p < 0.05, ** indicated that p < 0.01.
sMSCs have more DNA damage than yMSCs and an attenuated DNA repair ability
To evaluate the DNA repair ability of MSCs, the ability of the cells to repair oxidative DNA damage was determined using an alkaline comet assay. The extent of DNA damage in the form of single-strand breaks was measured as olive tail moment (OTM). As shown in Fig. 5A and B, DNA single-strand breaks were observed in both sMSCs and yMSCs 1 hour after treatment with H2O2. However, the OTM value of sMSCs was significantly higher than that of yMSCs (35.0±2.40 vs. 20.4±1.97, P<0.01), indicating that sMSCs have a higher amount of SSBs 1 hour following low-dose H2O2 treatment (25 μM), which could be due to a reduced antioxidant ability of sMSCs as shown in Fig. 3.
The DNA repair ability was attenuated in sMSCs. A. Alkaline comet assay of MSCs after 25 μM of H2O2treatmented. B. Quantification of OTM scores of A, 100 randomly chosen comets were analyzed. C. Alkaline comet assay of MSCs after 100 μM of H2O2 treatment. D. Quantification of OTM scores of C, 100 randomly chosen comets were analyzed. E. Neutral comet assay of MSCs after 30 μg/ml BLM treatment. F. Quantification of OTM scores of E, 100 randomly chosen comets were analyzed. G. Immunofluorescence microscopy of γH2AX foci in MSCs after 30 μg/ml BLM treatment. H. Quantification of γH2AX foci number in G, at least 100 randomly chosen nucleuses were analyzed and plotted. Data are presented as mean value±SD of three independent experiments. * indicated that p < 0.05, ** indicated that p < 0.01.
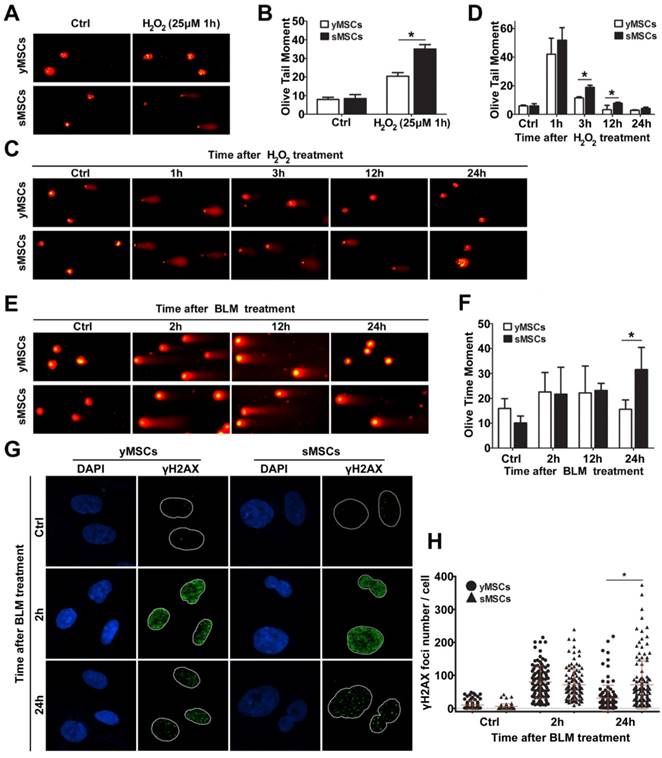
To evaluate the DNA repair ability in MSCs with an equal degree of damage, both sMSCs and yMSCs were treated with an overdose of H2O2 (100 μM). The comet assay was used in conjunction with OTM value determination for the MSCs at each time point (3h, 12h, and 24h post-treatment), where a decline in tail length represented the single-strand break DNA repair ability in each cell. As shown in Fig. 5 C and D, the decline rate of the OTM value in sMSCs was substantially less than that in yMSCs. The OTM value of yMSCs recovered to almost the normal level as soon as 12 hours after H2O2 exposure, whereas that of sMSCs remained greater than untreated cells up to 24 hours. These results indicate that the sMSCs have not only a decreased antioxidant ability but also an attenuated oxidative DNA repair ability, which may contribute to the replicative senescence of MSCs.
Two methods were used to compare the double-strand break repair ability of sMSCs and yMSCs. First, the repair kinetics was determined by neutral comet assay. The equal OTM value indicated that double-strand breaks were induced to the same extent in both cell types at 2 hours after BLM treatment, with little change at 12 hours after BLM treatment. However, at 24 hours after BLM treatment, the OTM value of yMSCs was almost recovered back to normal, whereas that of sMSC remained mostly unchanged (Fig. 5 E, F). This finding was confirmed by counting histone γH2AX foci, which are well-known markers of DSB. As shown in Fig. 5 G and H, 2 hours after BLM treatment, similar amounts of γH2AX foci were present in yMSCs and sMSCs, indicating equal levels of double-strand breaks in DNA. However, the clearance of γH2AX foci was substantially slower in sMSCs than in yMSCs as shown at 24 hours after BLM treatment, which suggests an attenuated double-strand break repair ability of sMSCs. These results indicated that the BLM-induced DNA damage was properly repaired in yMSCs but was sustained in sMSCs up to 24 hours after treatment.
Collectively, these results indicate that the DNA repair ability of both single- and double-strand breaks is greatly attenuated in sMSCs compared with yMSCs.
Discussion
In the present study, we aimed to assess the influence of aging on the cellular response to oxidative stress and DNA damage in BMSCs. This is the first study where the DDR of replicative senescent MSCs has been investigated in detail. The replicative senescent BM-MSCs were characterized and observed to be more sensitive to H2O2 and BLM treatment, both of which caused more DNA damage associated with attenuated DNA repair and anti-oxidative ability. Genes involved in DNA repair pathways and anti-oxidation were downregulated, including OGG1, XRCC1, Ku70, BRCA2, and XRCC4. In conclusion, stem cell senescence is associated with reduction in the DNA repair and anti-oxidative capacity.
Some evidence suggests that the complex process of aging is at least partially related to stem cell senescence. However, the mechanisms of stem cell aging remain to be defined. Primary human cells grown in culture are useful models for the analysis of aging, and such senescence in vitro is likely to mimic that occurring within human tissues [25]. It is generally agreed that the phenomenon of in vitro replicative senescence can recapitulate organism aging, and several studies have aimed to address the causes of cellular senescence in vitro [26, 27]. Although there may be multifactorial mechanisms that result in compromised stem cell functionality with advancing age, the accumulation of DNA damage within the stem cell compartment is likely to contribute to this process. Thus, comparison of the DNA damage response in normal and senescent MSCs could be used to determine the mechanism of senescence and aging. To date, some studies were conducted using a model of DNA damage that induces premature senescence or in age-related studies by comparison of MSCs between young and old donors [28-30]. This report is the first to characterize the DNA damage response in MSCs using a model of replicative senescence, which is considered to better mimic natural senescence in vivo and avoid the bias of cells obtained from different donors in an age-related study.
It is well known that oxidative stress plays a central role in aging and cell senescence [31]. Although cells in living organisms are continuously subjected to ROS, ROS generation is usually counteracted by enzymatic antioxidants. However, the regulation of intracellular ROS levels is critical for cell viability, as increases above normal concentrations lead to oxidative stress [32, 33], which induces various types of DNA damage mainly consisting of oxidized base damage, apurinic/apyrimidinic sites and DNA single-strand breaks [34, 35]. Accordingly, an efficient DNA repair system counteracts oxidative DNA damage and is a key factor in preventing cellular aging and senescence. Due to their long life span, stem cells are susceptible to DNA damage caused by both intrinsic and extrinsic oxidative stress, which might require specific protection from the long-term effects of ROS and oxidative damage. However, to date, few studies have investigated the promotion of aging by oxidative stress on human MSCs, and the mechanism by which oxidative stress induces stem cell aging is poorly understood.
Based on growing evidence, most normal stem cells can efficiently manage various types of DNA damage, including oxidative stress [36-38]. In this report, we postulated that accumulation of DNA damage in the aging process of MSCs is due to a defective DNA repair ability. With long-term cell culture in vitro, the features of antioxidant and DNA repair were characterized and compared between replicative senescence MSCs and their parental young MSCs. First, H2O2 -induced ROS in sMSCs was observed at substantially higher levels than that in yMSCs, which was accompanied by a lower expression of major antioxidant genes, including Mn-SOD, Cu/Zn-SOD, GPX, and CAT (Fig. 3). Moreover, with the comet assay, sMSCs displayed a substantially higher level of single-strand breaks 1 hour following low-dose treatment with H2O2 (25 µM) (Fig. 5A), which can also be explained by a reduced antioxidant ability of yMSCs shown in Fig. 3. Our results strongly suggest that sMSCs have attenuated antioxidant ability and might harbor more DNA damages. To address whether the greater level of oxidative DNA damage can be efficiently repaired or removed by DNA repair systems in sMSCs, DNA repair ability was evaluated using an overdose of H2O2 (100 μM) treatment to generate an equal extent of DNA damage in both cell types. Using the comet assay and gene expression analysis, the removal of ROS-induced single-strand breaks was shown to be decreased in sMSCs, which may partly be due to the lower expression or declined induction of BER DNA repair genes, including OGG1, and XRCC1 (Fig. 4 A, B). Consistent with the attenuated oxidative DNA repair ability, sMSCs were shown to be more sensitive to H2O2 with increased levels of apoptosis (Fig. 2). Collectively, these data indicate that the sMSCs have not only decreased antioxidant ability but also an attenuated oxidative DNA repair ability, both of which may contribute to the replicative senescence of MSCs.
Moreover, we attempted to address whether other specific DNA repair systems are also defective in sMSCs. Based on the fact that the DNA double-strand break is the most deleterious form of DNA damage, as well as the potential function of MSCs to promote tissue regeneration after anti-cancer drug therapy, BLM was chosen to induce DNA double-strand breaks in our study. This agent has been widely used for a variety of different cancers, including Hodgkin's lymphoma, testicular, ovarian and cervical cancers [39, 40]. Similar to the results in the analysis of oxidative DNA damage described above, sMSCs were shown to possess a defective DNA repair ability in response to double-strand breaks (Fig. 5 E to H), as they were more sensitive to BLM treatment as observed by enhanced apoptosis (Fig. 2 B, D). The impairment of DSB repair in sMSCs was further supported by the tail moment calculated from neutral comet assay after BLM exposure (Fig. 5 E, F) and sustained increase in γH2AX levels as determined by immunofluorescence detection (Fig. 5 G, H), which may at least partly be due to the reduced expression of HR or NHEJ genes, such as BRCA1, Ku70, and XRCC4 (Fig. 4). Nevertheless, we chose two representative types of DNA damage-as oxidative stress is mostly related to the natural replicative senescence process, and DSB being the most toxic form of DNA damage-to study the DNA repair ability in sMSCs. Together with the data above, we conclude that the maintenance of the adult stem cell genome through robust DNA repair is fundamental in the prevention of aging and disease; loss of DNA repair may contribute to the aging process by accelerating the accumulation of DNA damage induced by genetic dysfunction.
MSCs are one of the most promising candidates for cell-based therapy and tissue engineering for injured organs or tissues, and these applications depend on the expansion of cell culture in vitro. hMSCs in culture can undergo on average 40-60 divisions before they become senescent. This limited replicative capacity is also known as the “Hayflick limit” [41]. In recent years, accumulating data have demonstrated that MSCs from various animals undergo a spontaneous transformation during long-term culture, and escape from replicative senescence appears to be a crucial step in malignant transformation. The transformed MSCs are highly tumorigenic and are likely to be the tumor-initiating cells of sarcoma [8, 39, 42, 43]. Although there are contradictions regarding the spontaneous transformation of human MSCs during long-term culture [43-46], passage-dependent cancerous transformation of human MSCs that has occurred under carcinogenic hypoxia, oncogenic modification or by treating them with certain DNA-damaging carcinogens can be used to assess the associated mechanisms of senescence [47-49]. The safety of MSCs cannot be ensured with respect to a significant increase in chromosomal aberrations during in vitro expansion [10], and sequential transformation of MSCs is associated with increased radiosensitivity and reduced DNA repair capacity [50]. Thus, sMSCs have not only decreased antioxidant ability but also attenuated DNA repair ability, as shown in our study. Considering the more complicated environment in vivo, MSCs encountered various types of DNA damage more frequently than those that are expanded in vitro. Thus, based on the discovery of decreased DNA repair and antioxidant ability in replicative senescent MSCs, it is reasonable to propose that accumulation of numerous genetic and/or epigenetic alterations in adult stem/progenitor cells with advancing age may result in their immortalization and malignant transformation into highly leukemic or tumorigenic cancer-initiating cells and cancer initiation.
In conclusion, sMSCs are associated with a reduction in DNA repair and anti-oxidative capacity, partly due to the decreased expression of genes involved in DNA repair and anti-oxidation. These findings provide a better understanding of DNA damage responses in sMSCs and may lead to the development of better strategies for stem cell treatment and cancer therapy.
Acknowledgements
This work was supported by Grant (No. 81572772) from National Nature Science Foundation of China (NSFC) and Grant (No. cstc2014jcyjA10090) from the Nature Science Basic Research Foundation of Chongqing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD. et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143-7
2. Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276:71-4
3. Squillaro T, Peluso G, Galderisi U. Clinical Trials With Mesenchymal Stem Cells: An Update. Cell transplantation. 2016;25:829-48
4. Chen Y, Shao JZ, Xiang LX, Dong XJ, Zhang GR. Mesenchymal stem cells: a promising candidate in regenerative medicine. International Journal of Biochemistry & Cell Biology. 2008;40:815-20
5. Ksiazek K. A comprehensive review on mesenchymal stem cell growth and senescence. Rejuvenation research. 2009;12:105-16
6. Zhou YF, Bosch-Marce M, Okuyama H, Krishnamachary B, Kimura H, Zhang L. et al. Spontaneous transformation of cultured mouse bone marrow-derived stromal cells. Cancer research. 2006;66:10849-54
7. Miura M, Miura Y, Padilla-Nash HM, Molinolo AA, Fu B, Patel V. et al. Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem cells. 2006;24:1095-103
8. Tolar J, Nauta AJ, Osborn MJ, Panoskaltsis Mortari A, McElmurry RT, Bell S. et al. Sarcoma derived from cultured mesenchymal stem cells. Stem cells. 2007;25:371-9
9. Pan Q, Fouraschen SM, de Ruiter PE, Dinjens WN, Kwekkeboom J, Tilanus HW. et al. Detection of spontaneous tumorigenic transformation during culture expansion of human mesenchymal stromal cells. Experimental biology and medicine. 2014;239:105-15
10. Froelich K, Mickler J, Steusloff G, Technau A, Ramos Tirado M, Scherzed A. et al. Chromosomal aberrations and deoxyribonucleic acid single-strand breaks in adipose-derived stem cells during long-term expansion in vitro. Cytotherapy. 2013;15:767-81
11. Borgonovo T, Solarewicz MM, Vaz IM, Daga D, Rebelatto CL, Senegaglia AC. et al. Emergence of clonal chromosomal alterations during the mesenchymal stromal cell cultivation. Molecular cytogenetics. 2015;8:94
12. Rossiello F, Herbig U, Longhese MP, Fumagalli M, d'Adda di Fagagna F. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Current opinion in genetics & development. 2014;26:89-95
13. Gire V, Roux P, Wynford-Thomas D, Brondello JM, Dulic V. DNA damage checkpoint kinase Chk2 triggers replicative senescence. The EMBO journal. 2004;23:2554-63
14. d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nature reviews Cancer. 2008;8:512-22
15. Alessio N, Bohn W, Rauchberger V, Rizzolio F, Cipollaro M, Rosemann M. et al. Silencing of RB1 but not of RB2/P130 induces cellular senescence and impairs the differentiation potential of human mesenchymal stem cells. Cellular and molecular life sciences: CMLS. 2013;70:1637-51
16. Alessio N, Capasso S, Ferone A, Di Bernardo G, Cipollaro M, Casale F. et al. Misidentified Human Gene Functions with Mouse Models: The Case of the Retinoblastoma Gene Family in Senescence. Neoplasia. 2017;19:781-90
17. Collin G, Huna A, Warnier M, Flaman JM, Bernard D. Transcriptional repression of DNA repair genes is a hallmark and a cause of cellular senescence. Cell death & disease. 2018;9:259
18. Galderisi U, Helmbold H, Squillaro T, Alessio N, Komm N, Khadang B. et al. In vitro senescence of rat mesenchymal stem cells is accompanied by downregulation of stemness-related and DNA damage repair genes. Stem cells and development. 2009;18:1033-42
19. Nassour J, Martien S, Martin N, Deruy E, Tomellini E, Malaquin N. et al. Defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells. Nature communications. 2016;7:10399
20. Colter DC, Class R, DiGirolamo CM, Prockop DJ. Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3213-8
21. Colter DC, Class R, DiGirolamo CM, Prockop DJ. Rapid expansion of recycling stem cells in cultures of plastic-adherent cells from human bone marrow. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3213-8
22. Izadpanah R, Joswig T, Tsien F, Dufour J, Kirijan JC, Bunnell BA. Characterization of multipotent mesenchymal stem cells from the bone marrow of rhesus macaques. Stem cells and development. 2005;14:440-51
23. Barzilai A, Yamamoto K. DNA damage responses to oxidative stress. DNA repair. 2004;3:1109-15
24. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Molecular cell. 2012;47:497-510
25. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nature reviews Molecular cell biology. 2014;15:482-96
26. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nature reviews Molecular cell biology. 2007;8:729-40
27. Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nature reviews Molecular cell biology. 2007;8:703-13
28. Chen D, Cao G, Hastings T, Feng Y, Pei W, O'Horo C. et al. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. Journal of neurochemistry. 2002;81:1273-84
29. Tokalov SV, Gruner S, Schindler S, Wolf G, Baumann M, Abolmaali N. Age-related changes in the frequency of mesenchymal stem cells in the bone marrow of rats. Stem cells and development. 2007;16:439-46
30. Cabelof DC, Raffoul JJ, Ge Y, Van Remmen H, Matherly LH, Heydari AR. Age-related loss of the DNA repair response following exposure to oxidative stress. The journals of gerontology Series A, Biological sciences and medical sciences. 2006;61:427-34
31. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE. et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481-4
32. D'Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature reviews Molecular cell biology. 2007;8:813-24
33. Davies KJ. Oxidative stress: the paradox of aerobic life. Biochemical Society symposium. 1995;61:1-31
34. Cadet J, Ravanat JL, TavernaPorro M, Menoni H, Angelov D. Oxidatively generated complex DNA damage: tandem and clustered lesions. Cancer letters. 2012;327:5-15
35. Friedberg EC. DNA damage and repair. Nature. 2003;421:436-40
36. Blanpain C, Mohrin M, Sotiropoulou PA, Passegue E. DNA-damage response in tissue-specific and cancer stem cells. Cell stem cell. 2011;8:16-29
37. Oliver L, Hue E, Sery Q, Lafargue A, Pecqueur C, Paris F. et al. Differentiation-related response to DNA breaks in human mesenchymal stem cells. Stem cells. 2013;31:800-7
38. Valle-Prieto A, Conget PA. Human mesenchymal stem cells efficiently manage oxidative stress. Stem cells and development. 2010;19:1885-93
39. Albers P, Siener R, Krege S, Schmelz HU, Dieckmann KP, Heidenreich A. et al. Randomized phase III trial comparing retroperitoneal lymph node dissection with one course of bleomycin and etoposide plus cisplatin chemotherapy in the adjuvant treatment of clinical stage I Nonseminomatous testicular germ cell tumors: AUO trial AH 01/94 by the German Testicular Cancer Study Group. Journal of Clinical Oncology. 2008;26:2966-72
40. Murad AM, Triginelli SA, Ribalta JC. Phase II trial of bleomycin, ifosfamide, and carboplatin in metastatic cervical cancer. Journal of Clinical Oncology. 1994;12:55-9
41. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Experimental cell research. 1961;25:585-621
42. Zhou YF, Bosch-Marce M, Okuyama H, Krishnamachary B, Kimura H, Zhang L. et al. Spontaneous transformation of cultured mouse bone marrow-derived stromal cells. Cancer research. 2006;66:10849-54
43. Pan Q, Fouraschen SM, de Ruiter PE, Dinjens WN, Kwekkeboom J, Tilanus HW. et al. Detection of spontaneous tumorigenic transformation during culture expansion of human mesenchymal stromal cells. Experimental biology and medicine. 2014;239:105-15
44. Chen G, Yue A, Ruan Z, Yin Y, Wang R, Ren Y. et al. Human umbilical cord-derived mesenchymal stem cells do not undergo malignant transformation during long-term culturing in serum-free medium. PloS one. 2014;9:e98565
45. Aguilar S, Nye E, Chan J, Loebinger M, Spencer-Dene B, Fisk N. et al. Murine but not human mesenchymal stem cells generate osteosarcoma-like lesions in the lung. Stem cells. 2007;25:1586-94
46. Choumerianou DM, Dimitriou H, Perdikogianni C, Martimianaki G, Riminucci M, Kalmanti M. Study of oncogenic transformation in ex vivo expanded mesenchymal cells, from paediatric bone marrow. Cell proliferation. 2008;41:909-22
47. Shima Y, Okamoto T, Aoyama T, Yasura K, Ishibe T, Nishijo K. et al. In vitro transformation of mesenchymal stem cells by oncogenic H-rasVal12. Biochemical and biophysical research communications. 2007;353:60-6
48. Tang Q, Chen Q, Lai X, Liu S, Chen Y, Zheng Z. et al. Malignant transformation potentials of human umbilical cord mesenchymal stem cells both spontaneously and via 3-methycholanthrene induction. PloS one. 2013;8:e81844
49. Crowder SW, Horton LW, Lee SH, McClain CM, Hawkins OE, Palmer AM. et al. Passage-dependent cancerous transformation of human mesenchymal stem cells under carcinogenic hypoxia. FASEB Journal. 2013;27:2788-98
50. Worku M, Fersht N, Martindale C, Funes JM, Short SC. Sequential transformation of mesenchymal stem cells is associated with increased radiosensitivity and reduced DNA repair capacity. Radiation research. 2013;179:698-706
Author contact
![]() Corresponding author: Tel: 86-023-68754189; Fax: +86 23 68753260; E-mail: jinyangedu.cn
Corresponding author: Tel: 86-023-68754189; Fax: +86 23 68753260; E-mail: jinyangedu.cn