Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(6):595-602. doi:10.7150/ijms.23786 This issue Cite
Research Paper
Lipopolysaccharide Mediates the Destruction of Intercellular Tight Junction among Renal Tubular Epithelial Cells via RhoT1/SMAD-4/JAM-3 Pathway
Shixiang Zheng1,2 ![]() , Zhuoyong Lin3, Zhongwei Liu4, Yipeng Liu5, Wenwei Wu1
, Zhuoyong Lin3, Zhongwei Liu4, Yipeng Liu5, Wenwei Wu1 ![]()
1. Division of Critical Care Medicine, Union Hospital of Fujian Medical University, Fuzhou, Fujian, China 350001
2. Department of Vascular Surgery, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115
3. Deparment of Anesthesiology, Fujian Renmin Hospital, Fuzhou, Fujian, China 350001
4. Department of Cardiology, Shaanxi Provincial People's Hospital, Xi'an, Shaanxi, China 710068
5. Department of Nephrology, Qianfoshan Hospital, Shandong University, Jinan, Shandong, China 250014
Received 2017-11-10; Accepted 2018-3-2; Published 2018-3-14
Abstract
Background: The morbidity of sepsis induced acute kidney injury remains unacceptable high and the mechanisms of that disease remains unclear. For urine backleak and intercellular tight junction among tubular epithelial cells (TECs) destruction often occur during sepsis induced acute kidney injury, we examined whether lipopolysaccharide could damage intercellular tight junction among TECs and associated mechanisms in our present study.
Methods: HK-2 cells were cultured, transfected with different SiRNAs and stimulated with LPS and PYR-41. Transepithelial Permeability Assay and Transepithelial Electrical Resistance Assay were used to evaluate intercellular tight junction destruction and Western Blot and Immunofluorescence were used to evaluate proteins expression.
Results: Transepithelial Permeability increased significantly (P<0.05) and Transepithelial Electrical Resistance reduced remarkably (P<0.05) of the monolayer TECs stimulated with LPS. The expression of JAM-3 and RhoT1 decreased significantly (P<0.05) in TECs stimulated with LPS, while the level of SMAD-4 increased significantly (P<0.05). Downregulation of the expression of SMAD-4 with RNA interference could increase the expression of JAM-3 in LPS treated TECs. Moreover, upregulation of RhoT1 level by decreased the degradation of RhoT1 could decrease the expression of SMAD-4 and increase the JAM-3 level in TECs treated with LPS, while downregulation of RhoT1 level with RNA interference had the opposite effects.
Conclusion: LPS mediates intercellular tight junction destruction among TECs and RhoT1/SMAD-4/JAM-3 is a pivotal pathway to mediate the phenomenon.
Introduction
Sepsis is one of the most crucial diseases in hospitalized patients which remains the primary cause of in-hospital mortality. More than 60% of sepsis diagnoses are attributed to adults 65 years of age and older [1]. Kidney is the most vulnerable organ during sepsis while the morbidity of sepsis induced acute kidney injury (S-AKI) remains unacceptably high among septic patients, especially in the ICU [2]. However, to date, except avoidance of fluid volume deficit and nephrotoxic agents may reduce the incidence of S-AKI, there is no effective therapy that may be able to prevent tissue injury and alter the outcome of S-AKI. Moreover, it has been shown that more than half of patients with S-AKI progressed to chronic kidney disease (CKD), even to end stage renal disease (ESRD) especially in patients with CKD [3,4]. Therefore, understandingthe mechanisms that lead to AKI during sepsis become extremely important. To date, Studies have confirmed that inflammation plays the pivotal role in the occurrence of AKI [5,6] and that tubular backleak is one of the major phenomenon in kidney disease [7,8]. However, the exact mechanisms of tubular backleak during S-AKI remain unclear.
Gram-negative bacteria are the most common cause of sepsis while Lipopolysaccharide (LPS) plays the key role in this type of sepsis. Interacting of LPS with kinds of cells by different receptors such as TLR4 will cause cell and organ dysfunction which become one of the major mechanisms of sepsis induced organ failure [9,10]. Renal tubular epithelial cell (TEC) is one of most important inherent cells in kidney for maintainenance of normal renal function. The intercellular tight junction between TECs is of a pivotal factor in maintaining the integrity and function of renal tubules. Junctional adhesion molecule 3 (JAM-3) is a member of an immunoglobulin subfamily containing JAM-1, JAM-2, JAM-3, JAM-4, ESAM and CAR which is unique in terms of its broad expression and function [11], especially in maintaining the intercellular tight junction integrity [12,13]. JAM-3 is expressed by TECs and acts as the key role in the tight junction among the cells. Because the function of renal tubules decreases significantly, and tubular permeability increases remarkably during S-AKI, it is important to find out whether LPS stimulation could damage the tight junction among TECs by decreasing JAM-3 expression and associated mechanisms.
Here in our present study, we hypothesized that LPS could damage the tight junction among TECs by downregulating the expression of JAM-3. We examined our hypothesis in an in vitro system and studied relevant mechanisms.
Materials and Methods
Cell culture and treatments
Human renal epithelial cells (HK-2) were provided by American Type Culture Collection (ATCC) and cultured in Dulbecco's Modified Eagle's Medium/Nutrient Mixture F-12 (DMEM/F-12) supplemented with 10% heat-inactivated foetal calf serum (Gibco), 100U/ml penicillin G and 100ug/ml streptomycin at 37℃ in 5% CO2 in humidified incubator. Treatments for different groups were described in the results. Lipofectamine 2000 (Invitrogen) was used to perform SiRNA (RhoT SiRNA and SMAD-4 SiRNA, Qiagen) transfection according to the manufacturers' instruction.
Transepithelial Permeability Assay
The movement of Alexo488-albumin across the monolayer of cultured HK-2 cells grown on Transwell plates (0.4μm pore, Corning) were measured. Alexo488-albumin was added into the lower chamber of the Transwell plates at the concentration of 50μg/ml with or without LPS (Sigma) at the concentration of 0.5μg/ml. The movement of Alexo488-albumin to the upper chamber of the Transwell plates was monitored at 1 hour, 8 hours and 24 hours intervals with fluorescence microscopy and analyzed by Image J software.
Transepithelial Electrical Resistance Assay
HK-2 cells were cultured in Transwell Plates (0.4μm pore, Corning) until they achieved complete confluence. In LPS group, confluent monolayers of HK-2 cells were treated with LPS at the concentration of 0.5μg/ml and in the control group the HK-2 monolayers were cultured in the same medium without LPS. Transepithelial Electrical Resistance Assays were carried out with the Epithelial Volt/Ohmmeter (World Precision Instruments) and the resistance was presented in ohms divided by surface area (1.13cm2, Ω/cm2).
Western Blot Assay
Proteins expression were examined with Western Blot Assay. For cell derived samples, the total protein was extracted with RIPA buffer (150 mM sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecylsulfate, 50 mM Tris, and pH 8.0) containing a protease inhibitor cocktail (Sigma). Samples were centrifuged for 20 min at 4℃, 12000 rpm and mixed with loading buffer before being boiled at 70℃ for 10 min. Equal amounts of protein were separated by 10% SDS-gels (Life Technologies) and were transferred onto polyvinylidene fluoride membranes (GE Healthcare). The membranes then were cut into pieces based on the molecular weight of the target proteins and incubated with primary antibodies [RhoT1 rabbit monoclonal antibody, 1:500 (ProSci), SMAD-4 rabbit polyclonal antibody, 1:1000 (GeneTex), JAM-3 rabbit polyclonal antibody, 1:200 (Sigma)] at 4℃ for 12 hours. AlexaFluor 680/790-labeled goat anti-rabbit IgG antibody (1:10000, LI-COR Biosciences) was used as the secondary antibody. The blots were visualized by LI-COR Odyssey Infrared Imaging System (LI-COR Biosciences).
Immunofluorescence Assay
An immunofluorescence Assay was carried out as previously described. Briefly, the cell climbing films were fixed in 4% paraformaldehyde with 0.1% Triton X-100 for 30 min at 4℃. Subsequently, the films were incubated with the primary antibodies (SMAD-4 rabbit polyclonal antibody, 1:200 (GeneTex), RhoT1 rabbit monoclonal antibody, 1:100 (ProSci)) overnight at 4℃. The specimens were incubated with the selected secondary antibodies (FITC-conjugated goat anti-rabbit IgG antibody and TRITC-conjugated goat anti-rabbit IgG antibody) for 90 min in the dark. Hoechst 33342 was used to counterstain the nuclei. Microscopic images were taken and recorded by fluorescence microscopy or laser confocal microscopy (Olympus).
Statistical Analysis
Quantitative data were presented as the mean±SDs and Statistical analysis was performed using Graphpad Prism 6. Statistically significant differences in the mean values were assessed using Student's t-test or one-way analysis of variance with Tukey's multiple comparisons test for multiple comparisons. A P-value <0.05 was considered statistically significant.
Results
Effect of LPS on the intercellular tight junctions among TECs
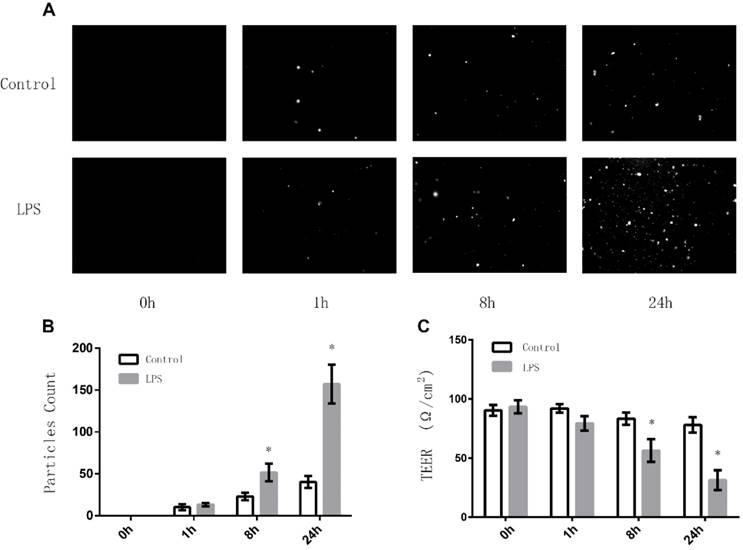
To investigate whether LPS stimulation can affect the intercellular tight junctions among TECs, we first examined the effects of LPS on the permeability of monolayer TECs (MLTECs) by preforming Transepithelial Permeability Assay with FITC-labeled Albumin. It can be demonstrated from the results that after an 8 hour treatment of LPS, the permeability of MLTECs increased significantly and continued to increase in a time dependent manner (Fig. 1A/B). Moreover, to further confirmour results, we also carried out Transepithelial Electrical Resistance (TEER) detection. As shown in figure 1, after an 8 hours LPS stimulation, the TEER level of the LPS group decreased significantly compared to the control and continue to decrease in a time dependent manner (Fig. 1C). All the results above demonstrated that LPS stimulation can disrupt the intercellular junctions among TECs.
Effects of LPS stimulation on Transepithelial Permeability Assay and TEER of MLTECs. (A) Representative fluorescent results of Transepithelial Permeability Assay in both LPS and Control groups (h: hours). (B) Statistical analysis of Transepithelial Permeability Assay results (*: P<0.05 vs. Control). (C) TEER results of both LPS and Control groups (*: P<0.05 vs. Control).
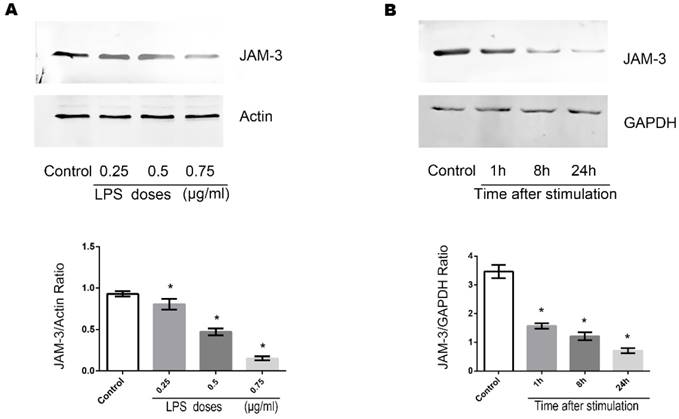
Effects of LPS on the expression of JAM-3 in TECs
JAM-3 is one of the most important intercellular adherent molecules expressed by TECs which contributes to the intercellular junctions. To further investigate the mechanisms through which LPS disrupts the junctions among TECs, we carried out Western Blot to determine the expression of JAM-3 in TECs. Stimulation of TECS with varying doses of LPS (0.25ug/ml, 0.5ug/ml and 0.75ug/ml) for 8 hours caused the expression of JAM-3 in TECs to decrease significantly in a dose dependent manner (Fig. 2A). Moreover, when TECs were stimulated with LPS at the concentration of 0.5ug/ml for various times (1 hour, 8 hours and 24 hours), the expression of JAM-3 also decreased significantly in a time dependent manner (Fig. 2B). All the results above indicated that LPS can down-regulate the expression of JAM-3 in TECs.
Effects of LPS stimulation on JAM-3 expression of TECs. (A) Representative Western Blotting results of JAM-3 expression levels of TECs stimulated with LPS at different concentrations for 8 hours (*: P<0.05 vs. Control). (B) Representative Western Blotting results of JAM-3 expression levels of TECs stimulated with LPS at the concentration of 0.5μg/ml for different hours (*: P<0.05 vs. Control).
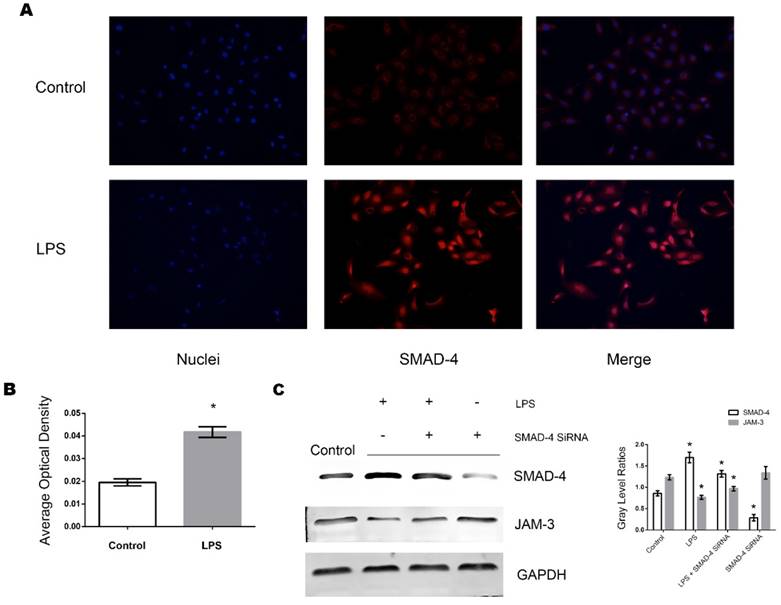
Role of SMAD-4 in the LPS induced downregulation of JAM-3 in TECs
SMAD-4 has been identified as a pivotal factor in the TGF-β pathway and has been shown to be upregulated in TECs in a variety of pathogenic processes including inflammation. We further investigated whether SMAD-4 was involved in the JAM-3 downregulation progress induced by LPS stimulation. It can be demonstrated from the immunofluorescent results that when stimulated with LPS (0.5ug/ml, 8 hours), the expression of SMAD-4 in TECs increased significantly and SMAD-4 was translocated into the nuclei (Fig. 3A, Fig. 3B). Furthermore, with Western Blot analysis, it can be demonstrated from the results that the expression of JAM-3 was significantly increased while the expression of SMAD-4 was downregulated by SMAD-4 SiRNA in TECs, and the JAM-3 down-regulation mediated by LPS stimulation was significantly reduced in the TECs transfected with SMAD-4 SiRNA (Fig. 3C). The results above demonstrated that SMAD-4 was a principal factor which mediated LPS induced JAM-3 downregulation in TECs.
Role of SMAD-4 in LPS induced JAM-3 downregulation. (A) Representative fluorescent results of SMAD-4 expression in TECs of both LPS and Control groups. (B) Statistical analysis of fluorescent results of SMAD-4 expression in TECs of LPS and Control groups (*: P<0.05 vs. Control). (C) Representative Western Blotting results of SMAD-4 and JAM-3 expressions in TECs in Control, LPS, LPS+SMAD-4 SiRNA and SMAD-4 SiRNA groups (*: P<0.05 vs. Control).
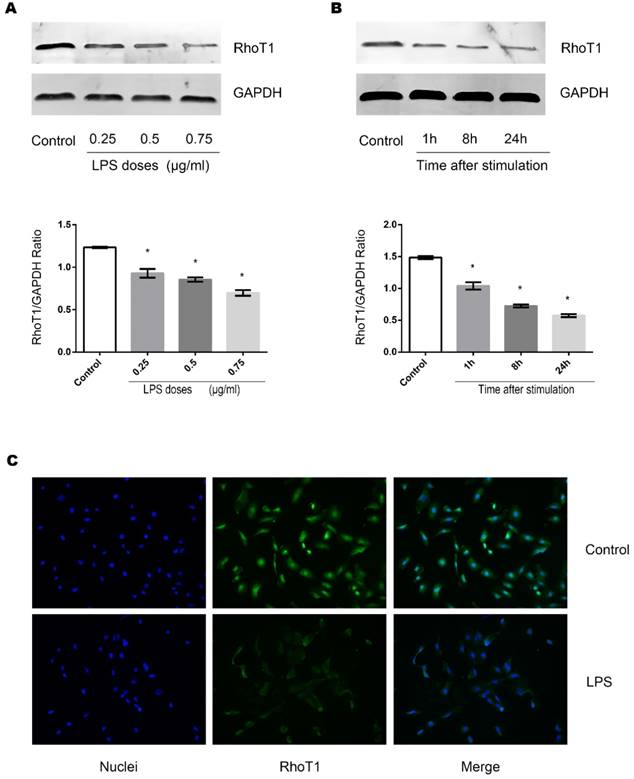
Effects of LPS on the expression of RhoT1 in TECs
RhoT1 has been proven as one of the up-stream regulators which affected the expression of SMAD-4. To further investigate whether LPS stimulation could alter the RhoT1 level in TECs, the expression of RhoT1 was measured by Western Blot and immunofluorescence. It can be indicated from the results that the expression of RhoT1 was decreased significantly when the TECs were stimulated by LPS and the decrement were in both time and dose dependent manners (Fig. 4A, Fig. 4B).
Effects of LPS stimulation on RhoT1 expression of TECs. (A) Representative Western Blotting results of RhoT1 expression levels of TECs stimulated with LPS at different concentrations for 8 hours (*: P<0.05 vs. Control). (B) Representative Western Blotting results of RhoT1 expression levels of TECs stimulated with LPS at the concentration of 0.5μg/ml for different hours (*: P<0.05 vs. Control). (C) Representative fluorescent results of RhoT1 expression in TECs of both LPS and Control groups.
Role of RhoT1 in LPS induced alteration of SMAD-4 and JAM-3 in TECs
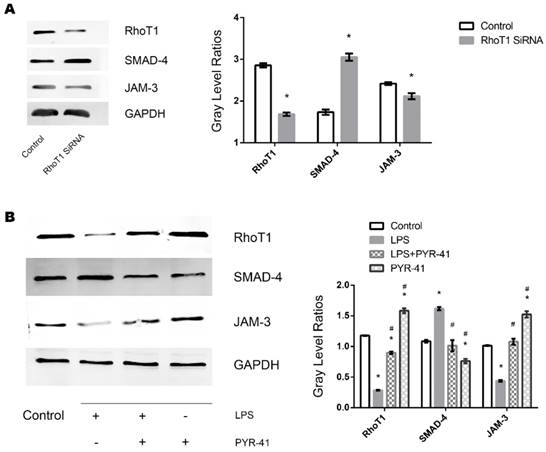
To further identify whether downregulation of RhoT1 could induce the upregulation of the expression of SMAD-4 expression, RhoT1 SiRNA was transfected into TECs and the relevant protein expression patterns were measured by Western Blot. As shown in the results, the expression of RhoT1 and JAM-3 were significantly reduced while the SMAD-4 levesl increased remarkably in the RhoT1 SiRNA group compared to the Control group (Fig. 5A). Since RhoT1 is degraded primarily through the ubiquitin proteasome pathway, the ubiquitin-activating enzyme (E1) inhibitor PYR-41 was used to determine whether upregulation of RhoT1 could downregulate the expression of SMAD-4 and promote the expression of JAM-3. By Western Blot it can be demonstrated that RhoT1 level decreased significantly in both the LPS and the LPS + PYR-41 groups but increased significantly in the PYR-41 group compared to Control, and that the RhoT1 levels of the LPS + PYR-41 and the PYR-41 group were significantly higher than that of the LPS group. The expression of SMAD-4 was significantly higher in the LPS group but significant lower in the PYR-41 group compared to the Control, and SMAD-4 levels of both the LPS + PYR-41 and the PYR-41 groups were significantly lower than that of the LPS group. The JAM-3 level of the PYR-41 group was significantly higher compared to the Control, and both the JAM-3 levels in the LPS + PYR-41 and the PYR-41 groups were significantly higher than that of the LPS group. At the same time, there were no significant difference between both expressions of SMAD-4 and JAM-3 in the Control group and in the LPS + PYR-41 group (Fig. 5B). All the results above indicated that the expressions of SMAD-4 and JAM-3 were regulated by RhoT1, and that downregulation of RhoT1 mediated the disruption of intercellular junctions via the SMAD-4/JAM-3 pathway.
Role of RhoT1 in LPS induced alteration of SMAD-4 and JAM-3 in TECs. (A) Representative Western Blotting results of Rho1, SMAD-4 and JAM-3 expressions in TECs of Control and RhoT1 SiRNA groups (*: P<0.05 vs. Control). (B) Representative Western Blotting results of Rho1, SMAD-4 and JAM-3 expressions in TECs of Control, LPS, LPS+PYR-41 and PYR-41 groups (*: P<0.05 vs. Control, #: P<0.05 vs. LPS group).
Discussion
S-AKI is a pivotal syndrome in ICU patients with poor prognosis and few treatment or prevention. It is crucial to explore the exact pathogenesis of S-AKI in order to discover further effective treatments for S-AKI. Previous studies indicated that urine backleak was commonly observed during AKI which acted as an important mechanism of that disease [14]. Furthermore, studies have indicated that when stimulated with the serum of sepsis patients, ZO-1 a crucial protein associated with intercellular tight junction decreased significantly [15]. Here in our present in vitro study, it was verified that when treated with LPS, albumin permeability of the MLTECs was increased remarkably and the TEER of MLTECs decreased significantly. Our results indicated that the intercellular tight junction between TECs was damaged by the stimulation of LPS. Our findings were consistent with previous studies and illustrated that LPS, the key factor in sepsis, can mediate the destruction of intercellular tight junction of TECs directly in vitro. We have then studied the possible molecular mechanisms that lead to this phenomenon.
JAM-3 is one of the key proteins which maintain the intercellular junctions among epitheliums and between epithelial cell and other types of cells [12,16]. The extracellular domains of JMA-3 interact with homophilic domains and also with integrins such as αMβ2, αXβ2 and αVβ3 [17]. Lamagra et al. demonstrated that JAM-3/JAM-2 binding was stronger than JAM-3 homophilic binding [18] and under oxidative stress, JAM-C is critically required for the survival of human cells [19]. Previous studies had also indicated that JAM-3 was expressed by epithelial cells and maintains the intercellular tight junction among them. However, there is few studies focus on the expression of JAM-3 in TECs during sepsis or TECs that are stimulated with LPS. Here in our present study, it has been found out that JAM-3 was expressed by TECs and the expressions were decreased significantly in both time dependent manner and dose dependent manner as stimulated with LPS. Our present results indicated that the LPS stimulation can down regulate the expression of JAM-3 in TECs to destruct the intercellular tight junction among the cells.
It is widely reconiged that SMAD-4 is a common SMADs molecule that plays a critical role in the signal transduction pathway of TGF-β by transducing SMAD-dependent signaling into nucleus. SMAD-4 regulates expressions of varies proteins as a nuclear transcription factor [20-22]. Previous studies indicated that SMAD-4 acts as a pivotal role in regulating the expression of molecules associated with intercellular junctions Previous studies also indicated that increasing SMAD-4 expression will result in downregulation of claudin-1 which is a crucial molecule in intercellular tight junction [23]. Here in our present study, it can be demonstrated from the results that when stimulated with LPS, the expression of SMAD-4 in TECs increased remarkably and SMAD-4 was transferred into nucleus. Moreover, with RNA interference technique, it was found out that the inhibition of SMAD-4 can significantly antagonize the down-regulation of JAM-3 in TECs induced by LPS. The results of our present study confirmed that SMAD-4 is an important molecule in the regulation of JAM-3 expression in TECs with LPS regulating JAM-3 expression through SMAD-4.
RhoT1 is an important regulatory protein which is expressed in various cells including TECs. Previous researches have indicated that RhoT1 plays a crucial role in maintaining the stability of mitochondria and regulating the distribution of mitochondria in cells [24,25]. Moreover, some studies also pointed out that RhoT1 could alter cell metabolism by regulating the transcriptions of various types of mRNA via SMAD-4 associated pathways [26]. RhoT1 is degraded by ubiquitin proteasome pathway in the cell, and its intracellular concentration is closely related to its synthesis level and the activation of ubiquitination pathway. Within our present study, it can be found out from the results that the expression of RhoT1 decreased significantly in TECs stimulated with LPS in both time and dose dependent manners. Furthermore, while inhibiting the expression of RhoT1 with RNA interference, SMAD-4 level was upregulated, and JAM-3 level was downregulated in TECs. This phenomenon is very similar with the changes of SMAD-4 and JAM-3 after LPS stimulation. For further inspection, we measured the expression of RhoT1 after LPS stimulation and the ubiquitin E1 inhibitor PYR-41 was used to inhibit the degradation of RhoT1. It was indicated from the results that the downregulation of RhoT1 was accompanied with remarkable upregulation of SMAD-4 and downregulation of JAM-3, and reducing the degradation of RhoT1 with PYR-41 reversed the process described above. Moreover, as RhoT1 level increased, the expression of SMAD-4 decreased remarkably in TECs treated with PYR-41. Our results confirmed that RhoT1 is an important upstream regulatory molecule that regulates the expression of SMAD-4, and its expression level played an upstream regulatory role in LPS-stimulated intercellular tight junction disruption.
We have recently found out in our previous researches that TECs actively participated the inflammatory storm during sepsis and that is a noble cause of the pathogenesis of S-AKI [6]. Here in our present study, we further discovered that LPS would directly induce intercellular tight junction destruction through RhoT1/SMAD-4/JAM-3 pathway when interacting with TECs. Although there may be other mechanisms that mediated the destruction of intercellular tight junction among TECs during sepsis, our study presented a noble mechanism leading to urine backleak and S-AKI. However, the present study has its limitations since it is an in vitro study. An in vivo study is required to further verify our results and to identify effective treatments for this pathogenic process.
In summary, the results presented here revealed that LPS can directly mediate intercellular tight junction destruction among TECs via the RhoT1/SMAD-4/JAM-3 pathway. S-AKI is a progressive clinical syndrome whose mechanism is poorly understood. It is also an independent risk factor for death among ICU patients. Our findings may provide a better understanding of the mechanisms of S-AKI and a basis for new treatment strategies.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 81701889) and the Start Up Foundation of Fujian Medical University (Grant No. 2016138).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rowe TA, McKoy JM. Sepsis in Older Adults. Infect Dis Clin North Am. 2017;4:731-742
2. Mohsenin V. Practical approach to detection and management of acute kidney injury in critically ill patient. J Intensive Care. 2017:57
3. He L, Wei Q, Liu J, Yi M, Liu Y, Liu H, Sun L, Peng Y, Liu F, Venkatachalam MA, Dong Z. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney International. 2017;5:1071-1083
4. O'Sullivan ED, Hughes J, Ferenbach DA. Renal Aging: Causes and Consequences. Journal of the American Society of Nephrology. 2017;2:407-420
5. Shi M, Zeng X, Guo F, Huang R, Feng Y, Ma L, Zhou L, Fu P. Anti-Inflammatory Pyranochalcone Derivative Attenuates LPS-Induced Acute Kidney Injury via Inhibiting TLR4/NF-kappaB Pathway. Molecules. 2017:10
6. Zheng S, Pan Y, Wang C, Liu Y, Shi M, Ding G. HMGB1 Turns Renal Tubular Epithelial Cells into Inflammatory Promoters by Interacting with TLR4 During Sepsis. J Interferon Cytokine Res. 2016;1:9-19
7. Eadon MT, Hack BK, Xu C, Ko B, Toback FG, Cunningham PN. Endotoxemia alters tight junction gene and protein expression in the kidney. Am J Physiol Renal Physiol. 2012;6:F821-F830
8. Singh P, Okusa MD. The role of tubuloglomerular feedback in the pathogenesis of acute kidney injury. Contributions to Nephrology. 2011:12-21
9. Savva A, Roger T. Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front Immunol. 2013:387
10. Qin X, Jiang X, Jiang X, Wang Y, Miao Z, He W, Yang G, Lv Z, Yu Y, Zheng Y. Micheliolide inhibits LPS-induced inflammatory response and protects mice from LPS challenge. Sci Rep. 2016:23240
11. Bradfield PF, Nourshargh S, Aurrand-Lions M, Imhof BA. JAM family and related proteins in leukocyte migration (Vestweber series). Arterioscler Thromb Vasc Biol. 2007;10:2104-2112
12. Mochida GH, Ganesh VS, Felie JM, Gleason D, Hill RS, Clapham KR, Rakiec D, Tan WH, Akawi N, Al-Saffar M, Partlow JN, Tinschert S, Barkovich AJ, Ali B, Al-Gazali L, Walsh CA. A homozygous mutation in the tight-junction protein JAM3 causes hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. American Journal of Human Genetics. 2010;6:882-889
13. Mandel I, Paperna T, Glass-Marmor L, Volkowich A, Badarny S, Schwartz I, Vardi P, Koren I, Miller A. Tight junction proteins expression and modulation in immune cells and multiple sclerosis. Journal of Cellular and Molecular Medicine. 2012;4:765-775
14. Kwon O, Nelson WJ, Sibley R, Huie P, Scandling JD, Dafoe D, Alfrey E, Myers BD. Backleak, tight junctions, and cell- cell adhesion in postischemic injury to the renal allograft. Journal of Clinical Investigation. 1998;10:2054-2064
15. Cantaluppi V, Weber V, Lauritano C, Figliolini F, Beltramo S, Biancone L, De Cal M, Cruz D, Ronco C, Segoloni GP, Tetta C, Camussi G. Protective effect of resin adsorption on septic plasma-induced tubular injury. Critical Care. 2010;1:R4
16. Hintermann E, Bayer M, Ehser J, Aurrand-Lions M, Pfeilschifter JM, Imhof BA, Christen U. Murine junctional adhesion molecules JAM-B and JAM-C mediate endothelial and stellate cell interactions during hepatic fibrosis. Cell Adh Migr. 2016;4:419-433
17. Garrido-Urbani S, Bradfield PF, Imhof BA. Tight junction dynamics: the role of junctional adhesion molecules (JAMs). Cell Tissue Res. 2014;355:701-15
18. Arcangeli ML, Frontera V, Bardin F, Thomassin J, Chetaille B, Adams S, Adams RH, Aurrand-Lions M. The Junctional Adhesion Molecule-B regulates JAM-Cdependent melanoma cell metastasis. FEBS Lett. 2012;586:4046-51
19. Jia X. et al. JAM-C maintains VEGR2 expression to promote retinal pigment epithelium cell survival under oxidative stress. Thromb Haemost. 2017;117(4):750-757
20. Chaikuad A, Bullock AN. Structural Basis of Intracellular TGF-beta Signaling: Receptors and Smads. Cold Spring Harb Perspect Biol. 2016:11
21. Ahmed S, Bradshaw AD, Gera S, Dewan MZ, Xu R. The TGF-beta/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J Clin Med. 2017:1
22. Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, Crystal RG, de Herreros AG, Moustakas A, Pettersson RF, Fuxe J. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nature Cell Biology. 2009;8:943-950
23. Shiou SR, Singh AB, Moorthy K, Datta PK, Washington MK, Beauchamp RD, Dhawan P. Smad4 regulates claudin-1 expression in a transforming growth factor-beta-independent manner in colon cancer cells. Cancer Research. 2007;4:1571-1579
24. Tada H, Taira Y, Morichika K, Kinoshita T. Mitochondrial trafficking through Rhot1 is involved in the aggregation of germinal granule components during primordial germ cell formation in Xenopus embryos. Development Growth & Differentiation. 2016
25. Shlevkov E, Kramer T, Schapansky J, LaVoie MJ, Schwarz TL. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc Natl Acad Sci U S A. 2016;41:E6097-E6106
26. Li Q, Yao L, Wei Y, Geng S, He C, Jiang H. Role of RHOT1 on migration and proliferation of pancreatic cancer. American Journal of Cancer Research. 2015;4:1460-1470
Author contact
![]() Corresponding authors: Shixiang Zheng, Division of Critical Care Medicine, Union Hospital of Fujian Medical University, Fuzhou, Fujian, China 350001. E-mail: Shawnsxzhengcom or szheng5harvard.edu And Wenwei Wu, Division of Critical Care Medicine, Union Hospital of Fujian Medical University, Fuzhou, Fujian, China 350001. athlaticnet
Corresponding authors: Shixiang Zheng, Division of Critical Care Medicine, Union Hospital of Fujian Medical University, Fuzhou, Fujian, China 350001. E-mail: Shawnsxzhengcom or szheng5harvard.edu And Wenwei Wu, Division of Critical Care Medicine, Union Hospital of Fujian Medical University, Fuzhou, Fujian, China 350001. athlaticnet