Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(6):557-563. doi:10.7150/ijms.23700 This issue Cite
Research Paper
Vascular calcification and left ventricular hypertrophy in hemodialysis patients: interrelationship and clinical impacts
Hyeon Seok Hwang1, Jung Sun Cho2, Yu Ah Hong1, Yoon Kyung Chang1, Suk Young Kim1, Seok Joon Shin1, Hye Eun Yoon1 ![]()
1. Division of Nephrology, Department of Internal Medicine, College of Medicine, The Catholic University of Korea
2. Division of Cardiology, Department of Internal Medicine, College of Medicine, The Catholic University of Korea
Received 2017-11-7; Accepted 2018-2-4; Published 2018-3-9
Abstract
Background: We examined the relationship and combined effect of vascular calcification (VC) and left ventricular hypertrophy (LVH) on deaths and cardiovascular events (CVEs) in hemodialysis (HD) patients.
Methods: Maintenance HD patients (n=341) were included. Echocardiography data and plain chest radiographs were used to assess LVH and aortic arch VC.
Results: VC was found in 100 patients (29.3%). LVH was more prevalent in patients with VC compared with those without VC (70% vs. 50.2%, P=0.001). VC was independently associated with a 2.42-fold increased risk of LVH (95% CI, 1.26-4.65). In multivariate analysis, compared with patients with neither VC nor LVH, the coexistence of VC and LVH was independently associated with CVE (HR, 2.01; 95% CI, 1.09-3.72), whereas VC or LVH alone was not. Patients with both VC and LVH had the highest risk for a composite event of deaths or CVE (HR, 1.88; 95% CI, 1.15-3.06). Significant synergistic interaction was observed between VC and LVH (P for interaction=0.039).
Conclusions: VC was independently associated with LVH. The coexistence of VC and LVH was associated with higher risk of deaths and CVEs than either factor alone. VC and LVH showed a synergistic interaction for the risk of deaths and CVEs.
Keywords: hemodialysis, cardiovascular event, death, left ventricular hypertrophy, vascular calcification
Introduction
End-stage renal disease (ESRD) patients requiring dialysis are at high risk for cardiovascular diseases [1], and cardiovascular disease is the major cause of death [2]. This is because the vascular system undergoes major structural and functional changes when renal function declines and dialysis is required [3]. Vascular calcification (VC) is a morphological marker of vascular pathologic changes [4], and an independent risk factor for deaths and cardiovascular events (CVEs) in ESRD patients [5-7]. VC is associated with arterial stiffness and functional and structural alterations in the heart, decreased coronary perfusion, and impaired renal and brain microcirculation [8, 9].
Left ventricular hypertrophy (LVH) is defined as an increase in left ventricular mass (LVM) consequent to increased wall thickness and it is a representative marker of cardiac structural pathology in ESRD patients. Multiple factors associated with decline in renal function increase the risk of LVH, including anemia, hypertension, hypervolemia, and disorders of mineral metabolism [2]. LVH has significant clinical significance since it is associated with adverse outcomes in dialysis patients. Previous reports showed that LVH and increase in LVM are associated with deaths and CVEs in dialysis patients [10-13].
There are few data on the interplay between VC and LVH in dialysis patients, while the vascular system and heart are tightly coupled. The study hypothesis was that VC is independently associated with LVH in hemodialysis (HD) patients, and that patients with both VC and LVH would have the highest risk for deaths and CVEs compared with those with VC or LVH alone.
Methods
Study population
We included ESRD patients who had received more than 1 month of HD treatment at Daejeon St. Mary's Hospital from February 2004 to November 2014. We included patients who were examined with plain chest radiographs for aortic arch VC and echocardiography for LVH. Subjects with the following criteria were excluded to avoid potential bias related to primary or secondary endpoint: current treatment for active infection, major surgery, overt signs of hemorrhage, acute heart failure, acute myocardial infarction, acute cerebral stroke, or an incomplete medical record. In total, 341 patients were enrolled. The sample was classified according to the presence or absence of VC, and each group was subdivided into two groups depending on the presence or absence of LVH.
Echocardiographic examination
Echocardiography was performed using a 15 MHz linear array transducer (Sequoia system, Acuson, Mountain View, CA, USA). M-mode and 2D measurements were conducted by trained sonographers in accord with methods recommended by the American Society of Echocardiography; cardiologists confirmed all echocardiographic results [14]. M-mode measurements included left ventricular end-diastolic diameter (LVEDD), left ventricular end-systolic diameter (LVESD), left ventricular posterior wall thickness at end-diastole (PWT), interventricular septal thickness at end-diastole (IVST), and aortic root diameter. Left ventricular ejection fraction (LVEF) and left atrial diameter (LAD) were determined from apical two- and four-chamber views by the Simpson's biplane formulae. LVM was calculated according to the formula: LVM = 0.80 × 1.04 × [(IVST + PWT + LVEDD)3 - LVEDD3] + 0.6 g, and then indexed for body surface area (LVMI). LVH was defined as LVMI >134 and >110 g/m2 for men and women, respectively [15, 16]. To estimate diastolic function, mitral inflow velocities were recorded at the apical four-chamber view using pulsed-wave Doppler. Peak early diastolic flow velocity (MV-E), peak late diastolic flow velocity (MV-A), and the ratio of E to A waves (E/A ratio) were measured [17]. From tissue Doppler imaging, septal mitral annular early peak velocity (E′) was determined, and the E/E′ ratio was calculated [18].
Data collection and definitions
Baseline demographics, risk factors for CVEs, laboratory data, antihypertensive medication, and HD procedure data were collected. VC of the aortic arch was identified using plain radiographs. Aortic arch calcification was observed by a single-blinded observer, and the total length of calcification was measured by adding the length of the separate linear calcific densities along the aortic arch [18]. A length of calcification >2 cm along the aortic arch was defined as VC. Body mass index (BMI) and body surface area (BSA) were calculated using the formulae:
BSA (m2) =
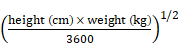
BMI (kg/m2) =
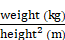
Pre-HD systolic and diastolic blood pressure (SBP and DBP) were calculated for each patient from the mean value for 1 month of HD treatment.
Outcome measures
The primary study endpoint was a composite of patient death or a CVE. A CVE was defined as the occurrence of coronary artery disease (coronary artery bypass surgery, percutaneous intervention, or myocardial infarction), heart failure, ventricular arrhythmia, sudden death, cerebrovascular accident (cerebral infarction, transient ischemic attack, or cerebral hemorrhage), or peripheral arterial disease (peripheral vascular revascularization, amputation, peripheral ulcer, or gangrene). The secondary endpoint was the association between VC and LVH.
Statistical analysis
Data are expressed as the mean ± standard deviation. Differences between two groups were identified using Student's t test. Categorical variables were compared using the chi-square test or Fisher's exact test. Binary logistic regression analysis was used to identify the independent association between VC and LVH. The Cox proportional hazards model was used to identify the independent variables related to the patient death or CVE. Multivariate models included the significantly associated parameters according to their weight in the univariate testing and clinically fundamental parameters. The confounders entered into the analysis were age (10-year increments), male, HD duration (1-month increments), BMI (1 kg/m2 increments), diabetes, previous CVE, mean SBP (10 mmHg increments), hemoglobin concentration (1 g/dL increments), serum levels of albumin (1 g/dL increments), total cholesterol (per 1 mg/dL increment), high-density lipoprotein cholesterol (HDL)-cholesterol (per 1 mg/dL increment), pre-HD SBP (per 10 mmHg increment), pulse pressure (per 10 mmHg increment), ultrafiltration volume (per 1 L increment), type of vascular access, and calcium channel blocker use. We conducted formal tests for interaction by including a VC-LVH interaction term in addition to the main effects to the fully adjusted models. The cumulative event rates were estimated using the Kaplan-Meier method and compared using the log-rank test. A P value of <0.05 was considered statistically significant. The statistical analyses were performed using SPSS software (version 20.0; SPSS, IBM Corp., Armonk, NY, USA).
Results
Baseline demographic characteristics and laboratory data
Table 1 shows the baseline sample characteristics. Among the 341 HD patients, 100 (29.3%) had VC at the aortic arch and LVH was more prevalent in patients with VC compared with those without VC (70% vs. 50.2%, respectively, P = 0.001). Among the patients with VC, those with LVH had higher mean pre-HD SBP and DBP, and pulse pressure compared with those without LVH. The use of beta-blockers and calcium channel blockers was more frequent in patients with LVH than in those without LVH. Among the patients without VC, those with LVH were less likely to be male and had lower hemoglobin levels and higher HDL-cholesterol level, mean pre-HD SBP and DBP, pulse pressure, and ultrafiltration volume, and used calcium channel blockers more frequently compared with those without LVH.
Baseline demographic and laboratory data of the study population
| VC | No VC | ||||||
|---|---|---|---|---|---|---|---|
| LVH (n = 70) | No LVH (n = 30) | P value | LVH (n = 121) | No LVH (n = 120) | P value | ||
| Age (years) | 68.1±10.0 | 68.9±10.3 | 0.710 | 54.0±12.5 | 55.8±15.3 | 0.333 | |
| Male (%) | 30 (42.9) | 15 (50.0) | 0.511 | 57 (47.1) | 75 (62.5) | 0.016 | |
| Body mass index (kg/m2) | 21.9±3.5 | 22.0±3.2 | 0.813 | 22.8±3.9 | 23.1±4.2 | 0.636 | |
| HD duration (years) | 2.6±4.6 | 2.7±5.6 | 0.273 | 14.7±40.2 | 15.2±41.5 | 0.922 | |
| Diabetes (%) | 5 (25.0) | 22 (59.5) | 0.726 | 52 (43.0) | 50 (41.7) | 0.837 | |
| Smoking (%) | 8 (11.4) | 3 (10.0) | 1.00 | 23 (19.0) | 26 (21.7) | 0.608 | |
| Previous CVE (%) | 6 (30.0) | 11 (29.7) | 0.092 | 33 (27.3) | 32 (26.7) | 0.916 | |
| Follow-up months | 27.6±25.8 | 25.8±22.4 | 0.741 | 35.3±34.9 | 39.6±33.2 | 0.326 | |
| Hemoglobin (g/dL) | 8.9±1.8 | 8.7±1.8 | 0.655 | 8.7±1.9 | 9.5±2.0 | 0.004 | |
| Albumin (g/dL) | 3.6±0.6 | 3.5±0.6 | 0.378 | 3.5±0.7 | 3.6±0.9 | 0.254 | |
| Total cholesterol (mg/dL) | 153.0±42.9 | 139.5±32.8 | 0.097 | 167.5±56.2 | 156.8±50.7 | 0.122 | |
| HDL-cholesterol (mg/dL) | 41.7±13.5 | 41.6±16.5 | 0.963 | 43.0±15.2 | 38.7±15.7 | 0.043 | |
| LDL-cholesterol (mg/dL) | 82.4±34.0 | 80.0±26.9 | 0.698 | 95.0±49.3 | 91.2±44.5 | 0.563 | |
| Calcium (mg/dL) | 8.9±1.2 | 8.6±1.1 | 0.362 | 8.3±1.4 | 8.5±1.4 | 0.210 | |
| Phosphorus (mg/dL) | 4.7±2.3 | 4.5±1.7 | 0.662 | 5.4±2.5 | 5.2±2.3 | 0.513 | |
| Intact PTH (pg/mL) | 143±147 | 183±353 | 0.438 | 186±187 | 201±264 | 0.622 | |
| Pre-HD SBP (mmHg) | 138.9±13.0 | 129.8±11.7 | 0.001 | 141.3±13.0 | 133.2±15.7 | <0.001 | |
| Pre-HD DBP (mmHg) | 82.7±5.5 | 78.9±5.4 | 0.001 | 84.5±5.5 | 80.8±7.2 | <0.001 | |
| Pulse pressure | 55.5±8.7 | 50.0±7.1 | 0.003 | 56.2±8.5 | 52.0±9.5 | <0.001 | |
| Ultrafiltration volume (L) | 1.9±0.9 | 1.8±0.9 | 0.585 | 2.0±0.9 | 1.8±0.9 | 0.016 | |
| Access type | |||||||
| Arteriovenous fistula | 47 (67.1) | 16 (54.3) | 0.246 | 77 (63.6) | 73 (60.8) | 0.603 | |
| Arteriovenous graft | 14 (20.0) | 11 (36.7) | 20 (16.5) | 17 (14.2) | |||
| Catheter | 9 (12.9) | 3 (10.0) | 24 (19.8) | 30 (25.0) | |||
| Anti-hypertensive medication | |||||||
| β-blocker | 33 (47.1) | 7 (23.3) | 0.026 | 43 (35.5) | 39 (32.5) | 0.619 | |
| Calcium channel blocker | 44 (62.9) | 9 (30.0) | 0.003 | 73 (60.3) | 49 (40.8) | 0.002 | |
| Renin-angiotensin system blocker | 37 (52.9) | 15 (50.0) | 0.793 | 63 (52.1) | 60 (50.0) | 0.748 | |
Abbreviations: BMI, body mass index; CVE, cardiovascular event; HD, hemodialysis; HDL-cholesterol, high-density lipoprotein cholesterol; LDL-cholesterol, low-density lipoprotein cholesterol; LVH, left ventricular hypertrophy; SBP, sytolic blood pressure; DBP, diastolic blood pressure; VC, vascular calcification.
Relationship between VC and LVH
Echocardiographic parameters were compared between patients with VC and those without VC (Table 2). Patients with VC had higher LVMI, LAD, and E/E′ compared with those without VC.
Comparison of echocardiographic measurements based on the status of VC
| VC (n = 100) | No VC (n = 241) | P value | |
|---|---|---|---|
| LVEDD (mm) | 49.9±6.6 | 49.2±6.5 | 0.429 |
| LVESD (mm) | 31.6±6.1 | 31.8±6.8 | 0.780 |
| PWT (mm) | 10.9±1.6 | 10.9±2.1 | 0.895 |
| IVST (mm) | 11.0±1.9 | 10.9±2.2 | 0.723 |
| LVMI (g/m2) | 136.0±35.4 | 126.0±40.6 | 0.032 |
| LVEF (%) | 62.7±9.3 | 60.5±10.8 | 0.079 |
| LAD (mm) | 38.9±7.5 | 37.0±7.1 | 0.032 |
| MV-E (m/sec) | 0.85±0.52 | 0.74±0.46 | 0.090 |
| MV-A (m/sec) | 1.01±0.20 | 0.95±0.24 | 0.055 |
| E/A ratio | 0.90±1.03 | 0.84±0.44 | 0.652 |
| E/E` ratio | 12.9±6.2 | 11.1±5.4 | 0.022 |
Abbreviations: E/A ratio, ratio of E to A waves; E′, septal mitral annular early peak velocity; IVST, interventricular septal thickness at end-diastole; LAD, left atrial diameter; LVEF, left ventricular ejection fraction; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; LVMI, left ventricular mass index; MV-E, peak early diastolic flow velocity; MV-A, peak late diastolic flow velocity; PWT, left ventricular posterior wall thickness at end-diastole.
Table 3 shows the determinants of LVH. In the univariate analysis, VC was significantly associated with LVH (odds ratio [OR], 2.31; 95% confidence interval [CI], 1.41-3.80; P = 0.001). Female gender, levels of hemoglobin, pre-HD SBP, pulse pressure, ultrafiltration volume, and use of calcium channel blockers were related to LVH. In the multivariate analysis, VC was independently associated with LVH (OR, 2.42; 95% CI, 1.26-4.65; P = 0.008). Female gender, levels of hemoglobin and pre-HD SBP, ultrafiltration volume, and use of calcium channel blocker were also independent determinants of LVH.
Logistic regression on the determinant factors of LVH
| Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|
| OR (95% CI) | P value | OR (95% CI) | P value | ||
| Age (per 10 years increment) | 1.02 (0.98, 1.18) | 0.808 | 0.997 (0.975, 1.018) | 0.761 | |
| Male (vs. female) | 0.56 (0.36, 0.86) | 0.008 | 0.45 (0.26, 0.78) | 0.004 | |
| HD duration (per 1 month increment) | 1.002 (0.997, 1.007) | 0.362 | 1.00 (0.99, 1.01) | 0.690 | |
| Diabetes (vs. absent) | 0.90 (0.59, 1.39) | 0.644 | 0.63 (0.36, 1.12) | 0.116 | |
| Previous CVE (vs. absent) | 1.32 (0.82, 2.13) | 0.260 | - | ||
| Hemoglobin (per 1g/dl increment) | 0.87 (0.77, 0.98) | 0.017 | 0.86 (0.75, 0.99) | 0.034 | |
| Serum albumin (per 1g/dl increment) | 0.92 (0.69, 1.22) | 0.553 | - | ||
| Serum calcium (per 1 mg/dL increment) | 0.98 (0.84, 1.15) | 0.822 | - | ||
| Serum phosphorus (per 1 mg/dL increment) | 1.02 (0.93, 1.11) | 0.756 | - | ||
| Intact PTH (per 1 pg/mL increment) | 0.99 (0.99, 1.00) | 0.296 | - | ||
| Total cholesterol (per 1 mg/dL increment) | 1.004 (0.999, 1.008) | 0.113 | 1.00 (0.99, 1.01) | 0.998 | |
| HDL-cholesterol (per 1 mg/dL increment) | 1.01 (1.00, 1.03) | 0.068 | 1.013 (0.996, 1.031) | 0.142 | |
| LDL-cholesterol (per 1 mg/dL increment) | 1.001 (0.996, 1.006) | 0.756 | - | ||
| Pre-HD SBP (per 10 mmHg increment) | 1.49 (1.27, 1.76) | <0.001 | 1.59 (1.06, 2.40) | 0.026 | |
| Pulse pressure (per 10 mmHg increment) | 1.72 (1.34, 2.20) | <0.001 | 0.93 (0.51, 1.68) | 0.929 | |
| Ultrafiltration volume (per 1 L increment) | 1.32 (1.03, 1.69) | 0.026 | 1.41 (1.02, 1.95) | 0.036 | |
| β-blocker use | 1.11 (0.95, 2.35) | 0.457 | - | ||
| Calcium channel blocker use | 2.51 (1.62, 3.89) | <0.001 | 2.31 (1.33, 4.01) | 0.003 | |
| Renin-angiotensin system blocker use | 1.10 (0.72, 1.69) | 0.666 | - | ||
| VC (vs. absent) | 2.31 (1.41, 3.80) | 0.001 | 2.42 (1.26, 4.65) | 0.008 | |
Abbreviations: CVE, cardiovascular event; HD, hemodialysis; HDL-cholesterol, high-density lipoprotein cholesterol; LDL-cholesterol, low-density lipoprotein cholesterol; LVH, left ventricular hypertrophy; SBP, systolic blood pressure; VC, vascular calcification.
Composite of deaths or CVEs
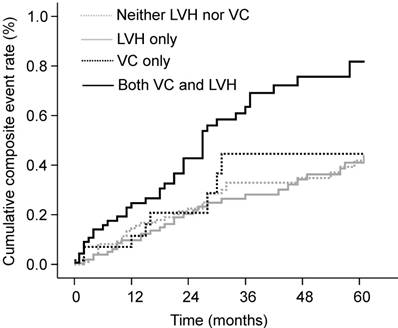
During follow-up, 65 deaths (19.1%) and 80 CVEs (23.5%) occurred. Patients with both VC and LVH showed the highest cumulative event rate for the composite of deaths or CVEs at 5-year follow-up (81.8%, P < 0.001; Fig. 1). Patients with either VC or LVH only had a 44.6% and 41.0% cumulative event rate, respectively, but these values were not significantly higher compared with those of patients with neither VC nor LVH (P = 0.722 and P = 0.623, respectively). Patients with both VC and LVH also had the highest cumulative event rate for mortality (P = 0.002) compared with 40.6%, 21.5%, and 16.5% for those with VC only, with LVH only, and with neither, respectively. The Kaplan-Meier event curves showed similar patterns for CVEs as those observed for composite event in the four groups: 71.0%, 38.6%, 35.4%, and 30.4% for patients with both VC and LVH, with VC only, with LVH only, and with neither, respectively (P = 0.005).
Cumulative rates of composite of deaths or CVEs according to the presence of VC and LVH. The cumulative event rate was highest in patients with both VC and LVH. CVE, cardiovascular event; LVH, left ventricular hypertrophy; VC, vascular calcification.
The observed incidence and hazard ratios (HRs) for patient deaths and CVEs are shown in Table 4. The incidence of composite of deaths or CVEs significantly differed according to the presence of VC and LVH (P < 0.001). In the univariate analysis, patients with both VC and LVH were significantly associated with the highest risk for composite of deaths or CVEs (HR, 2.63; 95% CI, 1.68-4.12; P < 0.001), mortality (HR, 2.55; 95% CI, 1.38-4.72; P = 0.003), and CVEs (HR, 2.46; 95% CI, 1.40-4.34; P = 0.002) compared with patients with neither VC nor LVH. In the multivariate Cox proportional hazards model, the coexistence of VC and LVH was significantly associated with the greatest risk for composite of deaths or CVEs (adjusted HR, 1.88; 95% CI, 1.15-3.06; P = 0.011), and there was a significant interaction between VC and LVH on the composite events (P = 0.039). Patients with both VC and LVH had the highest risk for CVEs (adjusted HR, 2.01, 95% CI, 1.09-3.72; P = 0.026) compared with patients with neither VC nor LVH.
Incidence and hazard ratios of deaths and CVE based on status of VC and LVH
| No. of events (%) | P | Unadjusted HR (95% CI) | Adjusted HR* (95% CI) | P value for interaction | |
|---|---|---|---|---|---|
| Composite of patient death and CVE | |||||
| Neither VC nor LVH | 39 (32.5) | <0.001 | Reference | Reference | 0.039 |
| VC only | 8 (26.7) | 1.16 (0.54-2.49) | 0.79 (0.34-1.83) | ||
| LVH only | 33 (27.3) | 0.89 (0.56-1.41) | 0.87 (0.53-1.41) | ||
| Both VC and LVH | 40 (57.1) | 2.63 (1.68-4.12) | 1.88 (1.15-3.06) | ||
| Patient deaths | |||||
| Neither VC nor LVH | 21 (17.5) | 0.055 | Reference | Reference | 0.418 |
| VC only | 6 (20.0) | 1.87 (0.75-4.69) | 1.18 (0.44-3.12) | ||
| LVH only | 17 (14.0) | 0.87 (0.46-1.66) | 0.84 (0.43-1.65) | ||
| Both VC and LVH | 21 (30.0) | 2.55 (1.38-4.72) | 1.60 (0.82-3.13) | ||
| CVE | |||||
| Neither VC nor LVH | 25 (20.8) | 0.124 | Reference | Reference | 0.380 |
| VC only | 6 (20.0) | 1.35 (0.55, 3.30) | 1.09 (0.40-2.93) | ||
| LVH only | 25 (20.7) | 1.06 (0.61-1.84) | 1.11 (0.62-1.99) | ||
| Both VC and LVH | 24(34.3) | 2.46 (1.40-4.34) | 2.01 (1.09-3.72) | ||
Abbreviations: BMI, body mass index; CVE, cardiovascular event; HD, hemodialysis; SBP, systolic blood pressure; VC, vascular calcification.
*Adjusted for age, BMI, diabetes, previous CVE, serum albumin level, serum total cholesterol level, preHD SBP, pulse pressure, ultrafiltration volume, vascular access type and calcium channel blocker use.
Discussion
This study showed that VC was independently associated with LVH and that the coexistence of VC and LVH was associated with a higher risk of deaths and CVEs than either factor alone. VC and LVH had a synergistic interaction on the risk of deaths and CVEs. These findings suggest that VC and LVH are closely related and that the coexistence of VC and LVH has a potent impact on mortality and cardiovascular outcomes.
Early-onset LVH is one of the most characteristic and prominent cardiac changes among patients with chronic kidney disease [3], which was associated with deaths and CVEs [10-13]. In ESRD patients, LVH is a multifactorial complication, caused by anemia, hypertension, hypervolemia, and disorders of mineral metabolism [2]. Our results were consistent with those showing that anemia and higher pre-HD SBP and ultrafiltration volume are independently associated with LVH and extended those findings by showing that LVH is more prevalent in patients with VC than in those without VC, and that VC is an independent determinant of LVH. These findings implicate the importance of VC for the development of LVH in HD patients.
In ESRD patients, both the intima and media thickness increase and VC occurs in the two areas of the vessel wall [3]. The inappropriate VC causes arterial fibroelastic thickening and loss of elastic fiber, which increases arterial stiffness and elevated pulse pressure [19-21]. Aortic stiffening associated with VC also increases cardiac afterload and arterial circumferential stress. Unfortunately, all of these structural and functional changes in vasculature promote LVH [9]. In addition, arterial dysfunction and LVH lead to increased filling pressure and limited diastolic filling, which leads to left ventricular (LV) diastolic stiffness [22]. Our results showed not only that VC is closely associated with LVH, but also that patients with VC had higher LAD and E/E′ ratio compared with those without VC. Therefore, we suggest that pathologic alterations in the arterial wall provide the background for the interaction of VC with LVH and diastolic dysfunction, and support the concept of vascular-ventricular coupling in HD patients [6, 7, 23].
In this study, neither the presence of VC without LVH nor the presence of LVH without VC was associated with mortality or CVEs. However, the coexistence of VC and LVH was an independent predictor for CVEs and composite of deaths or CVEs. These findings suggest that LVH may further affect HD patients with VC toward adverse outcomes. Since LVH causes limited diastolic LV filling and thereby reduces LV stroke volume [3, 22], LVH contributes to systolic LV dysfunction and heart failure. Accordingly, LVH increases the risk of ischemic injuries on the heart, brain, and peripheral arteries, and this was enhanced when VC or associated vascular dysfunction involved these organs [24]. Therefore, we suggest that the combined effect of VC and LVH on CVE risk should be considered to optimize the care of HD patients.
A synergistic interaction was found between VC and LVH on the risk of composite events. This finding suggests that VC and LVH exacerbate each other and there is a longitudinal relationship between VC and LVH in HD patients. VC alters the pulsatile dynamics in vasculature and consequently contributes to an increase in LV load and LVH [25]. LVH may also induce or aggravate VC in HD patients. HD patients with ultrafiltration results in systemic hemodynamic perturbation and myocardial perfusion impairment and are prone to falling in intradialytic hypotension [26, 27]. These episodes increase gut ischemia and lead to endotoxemia from the gut [28]. It has been shown that HD-induced systemic circulatory stress and recurrent regional ischemia result in increased endotoxin translocation from the gut and cause endotoxemia, and that this is related to markers of inflammation, malnutrition, cardiac injury, and reduced survival [29]. We speculate that patients with LVH may have greater circulatory stress during HD and consequently greater endotoxemia and more severe inflammation, all of which would contribute to VC, compared with those without LVH. In addition, endothelial biology is impaired in patients with LVH [30-32]. Given the close proximity of endothelial and smooth muscle cells in blood vessels, dysfunctional endothelial cells cause the development and progression of VC through the modulation of smooth muscle cells [33, 34].
Limitation
This study has some limitations. First, this was a single center study with a retrospective design. Second, VC was measured qualitatively using plain radiographs instead of computed tomography. Third, the pathophysiological mechanism of the interplay between VC and LVH was not proven.
Conclusions
VC was closely associated with LVH in HD patients and the coexistence of VC and LVH exposed patients to the highest risk of deaths and CVEs. In addition, there was a synergistic interaction between VC and LVH on adverse outcomes. Screenings and interventions for preventing or ameliorating VC or LVH are needed for ESRD patients at dialysis initiation. In patients with both VC and LVH, intensive management strategies should be continued to prevent future CVEs.
Abbreviations
end-stage renal disease: ESRD; vascular calcification: VC; cardiovascular event: CVE; left ventricular hypertrophy: LVH; left ventricular mass: LVM; hemodialysis: HD; left ventricular end-diastolic diameter: LVEDD; left ventricular end-systolic diameter: LVESD; left ventricular posterior wall thickness at end-diastole: PWT; interventricular septal thickness at end-diastole: IVST; left ventricular ejection fraction: LVEF; left atrial diameter: LAD; left ventricular mass index: LVMI; peak early diastolic flow velocity: MV-E; peak late diastolic flow velocity: MV-A; the ratio of E to A waves: E/A ratio; septal mitral annular early peak velocity: E′; body mass index: BMI; body surface area: BSA; systolic blood pressure: SBP; diastolic blood pressure: DBP; odds ratio: OR; confidence interval: CI; hazard ratio: HR
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2015R1A1A1A05001599), and through the NRF funded by by the Ministry of Science, ICT and future Planning (2014R1A1A3A04050919).
Ethics approval and consent to participate
Since this study was a retrospective one using clinical data, and it did not involve further invasive intervention, treatment, or costs to patients, the study received a consent exemption and it was approved by the Institutional Review Board of Daejeon St. Mary's Hospital (XC15RIMI0073O). The patient's record was de-identified and analyzed anonymously. This study was performed in accordance with the Declaration of Helsinki.
Authors' contribution
HSH and HEY contributed to conception and design and drafted the manuscript. HSH contributed to the analysis and interpretation. JSC, YAH, YKC, SYK, and SJS contributed to acquisition and interpretation. HEY contributed to interpretation. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kim H, Kim KH, Ahn SV. et al. Risk of major cardiovascular events among incident dialysis patients: A Korean national population-based study. Int J Cardiol. 2015;198:95-101
2. McCullough PA, Chan CT, Weinhandl ED, Burkart JM, Bakris GL. Intensive Hemodialysis, Left Ventricular Hypertrophy, and Cardiovascular Disease. Am J Kidney Dis. 2016;68:S5-s14
3. Wanner C, Amann K, Shoji T. The heart and vascular system in dialysis. Lancet. 2016;388:276-84
4. London GM. Cardiovascular calcifications in uremic patients: clinical impact on cardiovascular function. J Am Soc Nephrol. 2003;14:S305-9
5. London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731-40
6. Yoon HE, Chung S, Whang HC. et al. Abdominal aortic calcification is associated with diastolic dysfunction, mortality, and nonfatal cardiovascular events in maintenance hemodialysis patients. J Korean Med Sci. 2012;27:870-5
7. Yoon HE, Park BG, Hwang HS. et al. The prognostic value of abdominal aortic calcification in peritoneal dialysis patients. Int J Med Sci. 2013;10:617-23
8. London GM, Guerin AP, Marchais SJ. et al. Cardiac and arterial interactions in end-stage renal disease. Kidney Int. 1996;50:600-8
9. London G, Covic A, Goldsmith D. et al. Arterial aging and arterial disease: interplay between central hemodynamics, cardiac work, and organ flow-implications for CKD and cardiovascular disease. Kidney Int Suppl (2011). 2011;1:10-2
10. Silberberg JS, Barre PE, Prichard SS, Sniderman AD. Impact of left ventricular hypertrophy on survival in end-stage renal disease. Kidney Int. 1989;36:286-90
11. London GM, Pannier B, Guerin AP. et al. Alterations of left ventricular hypertrophy in and survival of patients receiving hemodialysis: follow-up of an interventional study. J Am Soc Nephrol. 2001;12:2759-67
12. Wang AY, Wang M, Woo J. et al. Inflammation, residual kidney function, and cardiac hypertrophy are interrelated and combine adversely to enhance mortality and cardiovascular death risk of peritoneal dialysis patients. J Am Soc Nephrol. 2004;15:2186-94
13. Zoccali C, Benedetto FA, Mallamaci F. et al. Left ventricular mass monitoring in the follow-up of dialysis patients: prognostic value of left ventricular hypertrophy progression. Kidney Int. 2004;65:1492-8
14. Sahn DJ, DeMaria A, Kisslo J, Weyman A. Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation. 1978;58:1072-83
15. Devereux RB, Alonso DR, Lutas EM. et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57:450-8
16. Glassock RJ, Pecoits-Filho R, Barberato SH. Left ventricular mass in chronic kidney disease and ESRD. Clin J Am Soc Nephrol. 2009;4(Suppl 1):S79-91
17. Rakowski H, Appleton C, Chan KL. et al. Canadian consensus recommendations for the measurement and reporting of diastolic dysfunction by echocardiography: from the Investigators of Consensus on Diastolic Dysfunction by Echocardiography. J Am Soc Echocardiogr. 1996;9:736-60
18. Nagueh SF, Middleton KJ, Kopelen HA, Zoghbi WA, Quinones MA. Doppler tissue imaging: a noninvasive technique for evaluation of left ventricular relaxation and estimation of filling pressures. J Am Coll Cardiol. 1997;30:1527-33
19. Schwarz U, Buzello M, Ritz E. et al. Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol Dial Transplant. 2000;15:218-23
20. London GM, Safar ME, Pannier B. Aortic Aging in ESRD: Structural, Hemodynamic, and Mortality Implications. J Am Soc Nephrol. 2016;27:1837-46
21. Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Renal Physiol. 2014;307:F891-900
22. Westermann D, Kasner M, Steendijk P. et al. Role of left ventricular stiffness in heart failure with normal ejection fraction. Circulation. 2008;117:2051-60
23. Fujiu A, Ogawa T, Matsuda N, Ando Y, Nitta K. Aortic arch calcification and arterial stiffness are independent factors for diastolic left ventricular dysfunction in chronic hemodialysis patients. Circ J. 2008;72:1768-72
24. Kim SY, Hong YA, Yoon HE. et al. Vascular calcification and intradialytic hypotension in hemodialysis patients: Clinical relevance and impact on morbidity and mortality. Int J Cardiol. 2016;217:156-60
25. Nitta K, Akiba T, Uchida K. et al. Left ventricular hypertrophy is associated with arterial stiffness and vascular calcification in hemodialysis patients. Hypertens Res. 2004;27:47-52
26. McIntyre CW, Burton JO, Selby NM. et al. Hemodialysis-induced cardiac dysfunction is associated with an acute reduction in global and segmental myocardial blood flow. Clin J Am Soc Nephrol. 2008;3:19-26
27. Schreiber MJ Jr. Clinical case-based approach to understanding intradialytic hypotension. Am J Kidney Dis. 2001;38:S37-47
28. Khanna A, Rossman JE, Fung HL, Caty MG. Intestinal and hemodynamic impairment following mesenteric ischemia/reperfusion. J Surg Res. 2001;99:114-9
29. McIntyre CW, Harrison LE, Eldehni MT. et al. Circulating endotoxemia: a novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6:133-41
30. Quttainah M, Al-Hejailan R, Saleh S. et al. Progression of matrixin and cardiokine expression patterns in an ovine model of heart failure and recovery. Int J Cardiol. 2015;186:77-89
31. Lee CW, Huang PH, Huang SS. et al. Decreased circulating endothelial progenitor cell levels and function in essential hypertensive patients with electrocardiographic left ventricular hypertrophy. Hypertens Res. 2011;34:999-1003
32. Cao C, Hu JX, Dong YF. et al. Association of Endothelial and Mild Renal Dysfunction With the Severity of Left Ventricular Hypertrophy in Hypertensive Patients. Am J Hypertens. 2016;29:501-8
33. Shroff R, Long DA, Shanahan C. Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol. 2013;24:179-89
34. Wu SY, Zhang BH, Pan CS. et al. Endothelin-1 is a potent regulator in vivo in vascular calcification and in vitro in calcification of vascular smooth muscle cells. Peptides. 2003;24:1149-56
Author contact
![]() Corresponding author: Hye Eun Yoon, MD, PhD, Division of Nephrology, Department of Internal Medicine, Incheon St. Mary's Hospital, College of Medicine, The Catholic University of Korea, 222 Banpo-daero, Seocho-gu,137-701, Republic of Korea. Phone: +82-32-280-5886, Fax: +82-32-280-5987
Corresponding author: Hye Eun Yoon, MD, PhD, Division of Nephrology, Department of Internal Medicine, Incheon St. Mary's Hospital, College of Medicine, The Catholic University of Korea, 222 Banpo-daero, Seocho-gu,137-701, Republic of Korea. Phone: +82-32-280-5886, Fax: +82-32-280-5987