Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(10):1002-1007. doi:10.7150/ijms.20809 This issue Cite
Research Paper
IL-36γ inhibits differentiation and induces inflammation of keratinocyte via Wnt signaling pathway in psoriasis
Wenming Wang, Xiaoling Yu, Chao Wu, Hongzhong Jin ![]()
Department of Dermatology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, China
Received 2017-5-1; Accepted 2017-7-6; Published 2017-8-18
Abstract
Psoriasis is a common inflammatory skin disease characterized by abnormal keratinocyte inflammation and differentiation that has a major impact on patients' quality of life. IL-36γ, a member of IL-36 cytokine family, is highly expressed in psoriasis and plays an important role in inflammation response and differentiation. However, the function of IL-36γ in differentiation and inflammation of keratinocyte in psoriasis has not been clearly identified. Thus, this study aimed to investigate the role of IL-36γ on differentiation and inflammation in HaCaT cells. HaCaT cells were divided into three groups: (1) Control group; (2) IL-36γ (100 ng/mL) group; (3) IL-36γ (100 ng/mL) + IWP-2 (1μM) group. Real time PCR was used to detect gene expression; the inflammation cytokines were examined by ELISA. We showed that treatment of HaCaT cells with IL-36γ significantly upregulated the expression levels of β-catenin, cyclin D1, and ki-67. IL-36γ also promoted the production of the inflammatory cytokines IFN-γ, IL-1β and IL-6, suppressed the expression of filaggrin, involucrin, keratin 1 and keratin 5. Meanwhile, we demonstrated the role of IWP-2, an inhibitor of the Wnt signaling pathway, in IL-36γ-treated HaCaT cells. Collectively, our findings suggest that IL-36γ inhibits differentiation and induces inflammation of keratinocyte via Wnt signaling pathway in psoriasis, this indicated that downregulation of IL-36γ may be a potential therapeutic option in psoriasis.
Keywords: Psoriasis, IL-36γ, Wnt signaling pathway, differentiation, inflammation
Introduction
Psoriasis is a common, chronic skin disease, affecting approximately 2%-3% of the population[1]. The disease is usually characterised with raised, well-demarcated, erythematous plaques with adherent silvery scales[2]. The scales are a result of hyperproliferation and aberrant differentiation of keratinocytes and infiltration of inflammatory cells, such as dendritic cells, macrophages, and T cells in the dermis and neutrophils, with some T cells in the epidermis[3,4]. The inflammatory infiltrate indicates that cytokines plays an important role in the pathogenesis of psoriasis, and interesting therapeutic targets could be identified, such as drugs targeting TNF-α, IL-17, IL-22, IL-23 and GM-CSF[1,5]. However, the exact mechanisms regulating these abnormal differentiation and immunological dysfunction remain largely unknown.
The cytokines of IL-1 family is a set of structurally related molecules that are pro-inflammatory mediators[6]. IL-1 family now consists of 11 cytokines and 9 cell surface receptors, many of which are overexpressed in both non-lesional and lesional psoriasis skin compared with healthy control skin[7]. The IL-36 family, formerly known as IL-1F cytokines, comprises the agonistic cytokines IL-36α (also called IL-1F6), IL-36β (also called IL-1F8), and IL-36γ (also called IL-1F9), as well as the antagonistic cytokine IL-36Ra (also called IL-1F5), which is related to the IL-1 family[8].
The IL-36s are primarily expressed by keratinocytes in a restricted manner. IL-36 expression in keratinocytes can be enhanced by IL-1α, TNF-α, and IL-17[9,10]. Furthermore, IL-36 was able to induce its own expression and various pro-inflammatory cytokines as well as augmented IL-17-mediated production of antibacterial peptides. In addition, IL-36 cytokines also have synergistic effects on the induction of antimicrobial peptides by IL-17A or TNFα. These all indicate that other pro-inflammatory cytokines and IL-36 can reinforce each other and cause similar responses in keratinocytes[11]. Especially, IL-36γ appears to have a central amplifying position in pro-inflammatory pathways at the interface between innate and adaptive immunity. Increased levels of IL-36 cytokines were observed in lesions of human psoriatic skin and confirmed in mouse models of psoriasis-like diseases. IL-36γ is highly expressed and specific for psoriatic inflammation but only weakly expressed in other inflammatory skin diseases, such as atopic dermatitis, lichen planus and eczematous diseases[9,11]. These features highlight IL-36γ may be a valuable future biomarker in psoriasis patients, however, the exact mechanisms responsible for the effect of IL-36γ on keratinocyte are still unclear.
The Wnt signaling pathway plays a vital role in a variety of biological activities, such as cell proliferation, cell-fate determination, and differentiation during adult homeostasis[12]. Previous studies have demonstrated membranous β-catenin expression was significantly decreased in the control group in comparison to the lesions of active psoriasis. Nuclear β-catenin expression was significantly increased in lesions of psoriasis in comparison to the control cases[13]. These suggest that β-catenin may play an important role in keratinocyte differentiation.
However, the biological role of IL-36γ in psoriasis pathogenesis, especially in keratinocyte inflammatory and differentiation, and the relationship between IL-36γ and Wnt signaling pathway is not clearly understood. In this study, we investigated the role of IL-36γ on the inflammatory and differentiation in HaCaT cells. We also evaluated the mechanism by which IL-36γ exerts its effect on differentiation and inflammatory processes.
Materials and Methods
Cell culture and Reagents
HaCaT cell line purchased from China Infrastructure of Cell Line Resources (Beijing, China) was maintained in MEM/EBSS (HyClone, Logan, UT, USA) with 10 % FBS (VACCA Biologics, Green Bay, WI, USA) and 1 % penicillin/streptomycin (HyClone, Logan, UT, USA). Cells were cultured at 37˚C in a humid incubator with 5% CO2. HaCaT cells in logarithmic phase were treated with 100 ng/ml recombinant human IL-36γ (R&D Systems, Minneapolis, MN, USA), as follow Wnt signaling pathway pharmacological inhibitor IWP-2 (Sigma, St. Louis, MO, USA) 1 μM for 30 min.
Quantitative Real-Time RT-PCR Analysis
For quantitative real-time RT-PCR analyses (qRT-PCR), total RNA was isolated from HaCaT cells by Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. RNA purity and quantity were measured on a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and 500 ng of total RNA was reverse-transcribed with the Goscript™ Reverse Transcription system (Thermo Fisher Scientific, Waltham, MA, USA). Real-time PCR was performed in Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using FastStart Universal SYBR Green (Roche, Pleasanton, CA, USA). Calculations were performed using the 2-ΔΔCt relative quantification method. Primers are listed in Table 1.
Sequences of primers used for PCR analysis
| Gene | Forward | Reverse |
|---|---|---|
| Involucrin | TCCTCCAGTCAATACCCATCAG | CAGCAGTCATGTGCTTTTCCT |
| Filaggrin | TGAAGCCTATGACACCACTGA | TCCCCTACGCTTTCTTGTCCT |
| Keratin 1 | GGCAGTTCCAGCGTGAAGTTTGTT | TTCTCCGGTAAGGCTGGGACAAAT |
| Keratin 5 | CAGAGCCACCTTCTGCGTCCTG | GCTGAAGCTACGACTGCCCCC |
| cyclin D1 | AACTACCTGGACCGCTTCCT | CCACTTGAGCTTGTTCACCA |
| Ki-67 | TGACAAGCCCACGACTGATGAGAA | CTTTGCCTGCTGATGGTGTTCGTT |
| β-catenin | AAAATGGCAGTGCGTTTAG | TTTGAAGGCAGTCTGTCGTA |
| GAPDH | CGGAGTCAACGGATTTGGTCGTAT | AGCCTTCTCCATGGTGGTGAAGAC |
ELISA Assay
Conditioned media collected from treated cells and untreated cells were centrifuged to remove cell debris, and the supernatants were collected. The IFN-γ, IL-1β and IL-6 concentrations in the supernatants were measured using ELISA kits (NeoBioScience Technology Company, Shenzhen, China). The optical density was measured with Thermo Scientific Varioskan Flash (Thermo Fisher Scientific, Waltham, MA, USA) at 450nm wavelength. The protein concentration was determined by comparing the relative absorbance of the samples with the standards.
Statistical Analysis
Data are represented as a mean ± SEM. Data were analyzed using SPSS 21.0 for Windows (SPSS, Chicago, IL, USA). Differences between experimental groups were assessed using the two-tailed unpaired Student's t test. P < 0.05 was considered as statistical significance.
Results
IL-36γ exposure-induced gene expression inhibited by IWP-2 pretreatment
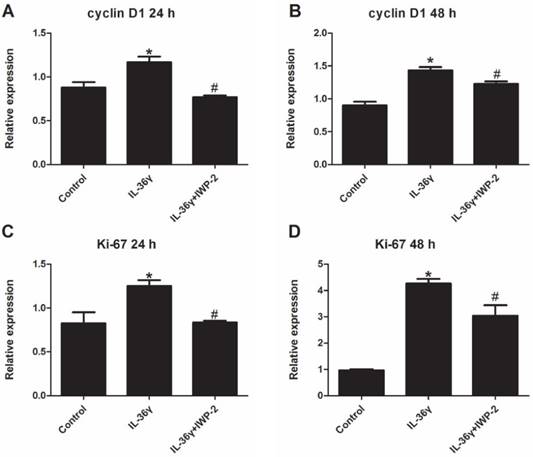
To determine the IL-36γ exposure-mediated upregulation of gene expression and the inhibitory effects of IWP-2, the cyclinD1 and ki-67 mRNA expression was detected at 24 h and 48 h after treatment. The cyclinD1 and ki-67 mRNA expression in the IL-36γ group increased significantly as compared to the Control group and decreased significantly by IWP-2 pretreatment at 24 h and 48 h (Fig.1A-D).
IWP-2 inhibits IL-36γ-induced expression of cyclinD1 and ki-67 in HaCaT cells. HaCaT cells were incubated with IL-36γ (100 ng/mL) for 24 h and 48 h in the presence or absence of IWP-2 (1 μM). IL-36γ exposure induced cyclinD1 and ki-67 upregulation. Treatment with IWP-2 significantly blunted IL-36γ induced cyclinD1 and ki-67 upregulation at mRNA level. ∗P < 0.05 vs. Control group; #P < 0.05 vs. IL-36γ group.
IL-36γ exposure-induced differentiation markers downregulation inhibited by IWP-2 pretreatment
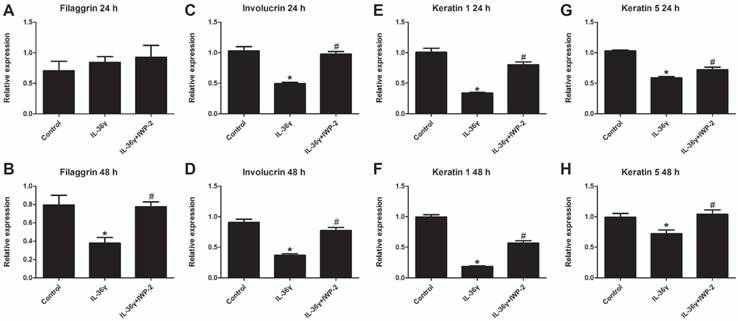
To determine the IL-36γ exposure-mediated downregulation of differentiation markers and the inhibitory effects of IWP-2, the filaggrin, involucrin, keratin 1 and keratin 5 mRNA expression was evaluated at 24 h and 48 h after treatment. The filaggrin mRNA expression in the IL-36γ group decreased significantly as compared to the Control group and increased significantly by IWP-2 pretreatment at 48 h (Fig.2A and B). The involucrin, keratin 1 and keratin 5 mRNA expression in the IL-36γ group decreased significantly as compared to the Control group and increased significantly by IWP-2 pretreatment at 24 h and 48 h (Fig.2C-H).
IWP-2 inhibits IL-36γ-induced downregulation of differentiation markers in HaCaT cells. Filaggrin, involucrin, keratin 1 and keratin 5 mRNA levels in HaCaT cells were assessed by real-time PCR. HaCaT cells were incubated with IL-36γ (100 ng/mL) for 24 h and 48 h in the presence or absence of IWP-2 (1 μM). ∗P < 0.05 vs. Control group; #P < 0.05 vs. IL-36γ group.
IL-36γ exposure-induced pro-inflammatory cytokines upregulation inhibited by IWP-2 pretreatment
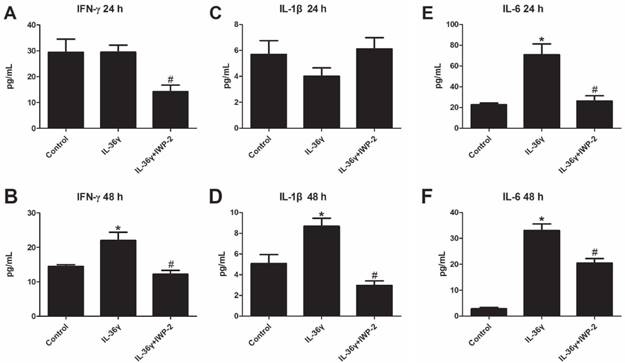
To explore the effects of IL-36γ on pro-inflammatory cytokines in HaCaT cells, ELISA analyses was used to detect the expression of IFN-γ, IL-1β and IL-6. We found that the IFN-γ and IL-1β expression was significantly increased after IL-36γ exposure and decreased significantly by IWP-2 pretreatment when compared with the Control group at 48 h(Fig.3A-D); the IL-6 expression was significantly increased after IL-36γ exposure and decreased significantly by IWP-2 pretreatment when compared with the Control group at 24h and 48 h(Fig.3E-F).
IWP-2 inhibits IL-36γ-induced upregulation of proinflammatory cytokines in HaCaT cells. IFN-γ, IL-1β and IL-6 levels of conditioned media were assessed by ELISA. Conditioned media were collected from HaCaT cells incubated with IL-36γ (100 ng/mL) for 24 h and 48 h in the presence or absence of IWP-2 (1 μM). ∗P < 0.05 vs. Control group; #P < 0.05 vs. IL-36γ group.
Wnt signaling pathway participated in the biological role of IL-36γ in HaCaT cells
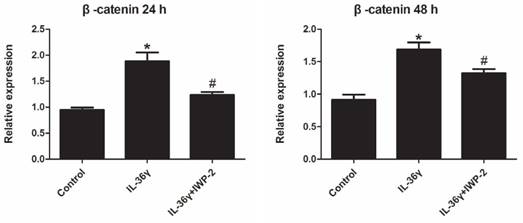
Expression levels of β-catenin in HaCaT cells were investigated using real time PCR.
Exposure to IL-36γ for 24 h and 48 h resulted in increase of β-catenin. Treatment with Wnt signaling pathway inhibitor IWP-2 blunted the increase expression of β-catenin (Fig.4).
IWP-2 inhibits IL-36γ-induced expression of β-catenin in HaCaT cells. HaCaT cells were incubated with IL-36γ (100 ng/mL) for 24 h and 48 h in the presence or absence of IWP-2 (1 μM). IL-36γ exposure induced β-catenin upregulation. Treatment with IWP-2 significantly blunted IL-36γ induced β-catenin upregulation at mRNA level. ∗P < 0.05 vs. Control group; #P < 0.05 vs. IL-36γ group.
Discussion
The present study shows that IL-36γ exposure induced β-catenin upregulation, differential inhibition, and pro-inflammation cytokines secretion. Nevertheless, the Wnt signaling pathway pharmacological inhibitor IWP-2 administration could suppress the β-catenin expression, improve differentiation of keratinocyte and inhibit pro-inflammation cytokines secretion. To our knowledge, this is the first report displaying the inhibitory effect of IWP-2 in HaCaT cells after IL-36γ exposure.
In recent years, major issues in elucidating the molecular mechanisms of psoriasis remain unresolved, including the autoimmune cause of the inflammatory process, the relevance of cutaneous versus systemic factors, and the role of genetic and environmental influences on the pathogenesis of the disease[14]. Previous studies have demonstrated that expression of IL-36γ is upregulated in colonic epithelial cells from patients with inflammatory bowel disease (IBD) [15,16], whereas in the lung, IL-36γ expression is strongly induced in various models of asthma can be produced by bronchial epithelium exposed to viral infection, smoke or inflammatory cytokines[17,18]. In the present study, we also found that a single exposure to IL-36γ impaired the normal function of HaCaT keratinocyte, suggesting that IL-36γ may play a role in the development of psoriasis skin lesion.
The epidermis is a self-renewing tissue, which functions to protect the interior of the body. Regular keratinocyte differentiation is crucial for the formation of an intact epidermal barrier. Epidermal barrier dysfunction and aberrant keratinocyte differentiation are involved in the pathophysiology of several skin diseases, such as psoriasis, atopic dermatitis, basal and squamous skin cancer[19]. Psoriasis is histologically characterized by increased thickness of the epidermis- acanthosis, expansion of the cornified layer- hyperkeratosis, retention of nuclei in the cornified layer- parakeratosis[20]. Besides their inflammatory aspects, full-blown psoriatic lesions show the distinctive signs of altered epidermal differentiation. The entire keratinization process in psoriasis is not completed because that generally accelerated keratinization is interrupted by premature cell death[21]. Previous studies have reported that the aberrant differentiation of keratinocytes that underlies the clinical appearance of psoriatic lesions. The early differentiation markers, such as small proline-rich proteins (SPRR), cystatin A and transglutaminase 1, are more conspicuously expressed in psoriasis, while the late differentiation markers, such as profilaggrin and loricrin, are down-regulated [21,22]. Investigations have confirmed that the abnormal differentiation of keratinocyte leads to the impairment of the skin barrier function. In our study, we found that treated HaCaT cells with IL-36γ can inhibit the differentiation of keratinocyte, lead to the downregulation of filaggrin, involucrin, keratin 1 and keratin 5 at mRNA levels. In addition, IWP-2 can block the effects of IL-36γ on HaCaT cells. These results suggest a major role for IL-36γ in abnormal differentiation of keratinocyte.
Skin inflammation that seen in psoriasis results from the multipartite interactions of, at least, KC, T cells, antigen presenting cells (APC), fibroblasts and endothelial cells. The hypothesis that a cytokine network in psoriasis proposed a central role of pro-inflammatory cytokines has been validated by the clinical success of anti-TNF and anti-IL-17A therapy in the treatment of psoriasis[23]. In psoriasis, the keratinocyte actively participates in a multicellular, multimolecular network coordinated by cytokines. Psoriatic keratinocytes are a rich source of antimicrobial peptides, including LL-37, β-defensins, and S100A7 (psoriasin), which show the effects of chemotaxis and shape immune-cell function[24,25]. They can response to dendritic cell and T-cell-derived cytokines, including interferons, TNF, interleukin-17, and so on. In turn, keratinocytes will produce pro-inflammatory cytokines (interleukin-1, interleukin-6, interleukin-8, TNF-α and so on)[1,11,26]. Many of the inflammatory mediators induced by IL-36 have been demonstrated to be over-expressed in psoriatic skin lesions and their continued action appears important in maintaining the lesional phenotype suggesting that IL-36 may contribute to the chemokine environment of inflamed skin[27]. Previous studies have suggested that increased expression of IL-36γ cytokines in in human psoriatic lesional skin as well as psoriasis-like mouse skin lesions[9]. However, there is little knowledge about the function of IL-36γ in the inflammatory response of keratinocytes in psoriasis. In our study, we found that treated HaCaT cells with IL-36γ can mimic the inflammatory process in psoriasis and found that IWP-2 suppressed the production of the inflammatory factors IL-1β, IL-6 and IFN-γ, implying a pro-inflammatory role of IL-36γ in HaCaT cells.
Wnt signaling pathway has an important role in embryonic development and homeostatic self-renewal in adult tissues, besides it exerts both anti-inflammatory and pro-inflammatory functions depending on the conditions and regulated through different mechanisms[12]. The β-catenin is the key regulatory factor in the Wnt signaling pathway activation. Previous studies have suggested that IL-22 promote bony proliferation in the paws of this experimental mouse model of spondyloarthropathy via Wnt signaling pathway [28,29]. In another report, the authors revealed that the β-catenin expression in synovium sample from patients with RA was significantly higher than that of trauma samples[30]. In a study focus on the role of β-catenin in the inhibition of epithelization and wound healing, did not find any nuclear β-catenin in normal epidermis. Decreased membrane staining and increased cytoplasm/nuclear β-catenin staining in the lesions of patients with active psoriasis have been reported[13]. The downregulation of membranous β-catenin in lesional psoriatic skin might be used as phenotypic marker of hyperprolifration of keratinocytes in psoriasis[13,31]. Thus, to improve the related mechanisms that involved in IL-36γ-regulated differentiation and inflammation, the Wnt signaling pathway is explored in the present study, we found that Wnt signaling pathway is a crucial pathway in IL-36γ-regulated differentiation and inflammation of HaCaT cells, suggesting that IL-36γ may play a critical role in suppressing the differentiation and encouraging inflammatory response by association with Wnt signaling pathway. However, additional studies are necessary to understand more clearly the role of IL-36γ in keratinocyte of psoriasis.
Our investigation has several limitations. First, as an inhibitor of Wnt production, IWP-2 may not special for canonical Wnt pathway or non-canonical Wnt pathway. Therefore, canonical Wnt pathway or non-canonical Wnt pathway, which is more important cannot be determined from this study. Second, we did not investigate the protein expression and cellular distribution of β-catenin. Further research on this issue is needed.
Overall, the present study showed that in HaCaT cells, IL-36γ can suppress the differentiation and promote the inflammatory response by the Wnt signaling pathway. These findings suggest that IL-36γ may represent potential targets for the treatment of psoriasis.
Acknowledgements
The present study was supported by grants from Capital Health Development Research Fund of China (2016-2-4018).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Baliwag J, Barnes DH, Johnston A. Cytokines in psoriasis. Cytokine. 2015;73:342-350
2. Tschachler E. Psoriasis: the epidermal component. Clin Dermatol. 2007;25:589-595
3. Christensen TE, Callis KP, Papenfuss J. et al. Observations of psoriasis in the absence of therapeutic intervention identifies two unappreciated morphologic variants, thin-plaque and thick-plaque psoriasis, and their associated phenotypes. J Invest Dermatol. 2006;126:2397-2403
4. Sticherling M. Psoriasis and autoimmunity. Autoimmun Rev. 2016;15:1167-1170
5. Gudjonsson JE, Johnston A, Ellis CN. Novel systemic drugs under investigation for the treatment of psoriasis. J Am Acad Dermatol. 2012;67:139-147
6. Dunn E, Sims JE, Nicklin MJ. et al. Annotating genes with potential roles in the immune system: six new members of the IL-1 family. Trends Immunol. 2001;22:533-536
7. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89-102
8. Johnston A, Xing X, Guzman AM. et al. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol. 2011;186:2613-2622
9. A.M. D'Erme, D. Wilsmann-Theis, J. Wagenpfeil, M, et al. IL-36gamma (IL-1F9) is a biomarker for psoriasis skin lesions. J Invest Dermatol. 2015;135:1025-1032
10. Tortola L, Rosenwald E, Abel B. et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. 2012;122:3965-3976
11. Carrier Y, Ma HL, Ramon HE. et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol. 2011;131:2428-2437
12. Ma B, Hottiger MO. Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during Inflammation. Front Immunol. 2016;7:378
13. El-Wahed Gaber MA, El-Halim Kandil MA. et al. Beta-catenin expression in psoriasis. Indian Dermatol Online J. 2015;6:13-16
14. Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866-873
15. Nishida A, Hidaka K, Kanda T. et al. Increased Expression of Interleukin-36, a Member of the Interleukin-1 Cytokine Family, in Inflammatory Bowel Disease. Inflamm Bowel Dis. 2016;22:303-314
16. Kanda T, Nishida A, Takahashi K. et al. Interleukin(IL)-36alpha and IL-36gamma Induce Proinflammatory Mediators from Human Colonic Subepithelial Myofibroblasts. Front Med (Lausanne). 2015;2:69
17. Friedrich M, Tillack C, Wollenberg A. et al. IL-36gamma sustains a proinflammatory self-amplifying loop with IL-17C in anti-TNF-induced psoriasiform skin lesions of patients with Crohn's disease. Inflamm Bowel Dis. 2014;20:1891-1901
18. Walsh PT, Fallon PG. The emergence of the IL-36 cytokine family as novel targets for inflammatory diseases. Ann N Y Acad Sci. 2016
19. Elsholz F, Harteneck C, Muller W. et al. Calcium-a central regulator of keratinocyte differentiation in health and disease. Eur J Dermatol. 2014;24:650-661
20. Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263-271
21. Iizuka H, Takahashi H, Honma M. et al. Unique keratinization process in psoriasis: late differentiation markers are abolished because of the premature cell death. J Dermatol. 2004;31:271-276
22. Iizuka H, Ishida-Yamamoto A, Honda H. Epidermal remodelling in psoriasis. Br J Dermatol. 1996;135:433-438
23. Deng Y, Chang C, Lu Q. The Inflammatory Response in Psoriasis: a Comprehensive Review. Clin Rev Allergy Immunol. 2016;50:377-389
24. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509
25. Bridgewood C, Stacey M, Alase A. et al. IL-36gamma has proinflammatory effects on human endothelial cells. Exp Dermatol. 2017;26:402-408
26. Vigne S, Palmer G, Lamacchia C. et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood. 2011;118:5813-5823
27. Towne JE, Sims JE. IL-36 in psoriasis. Curr Opin Pharmacol. 2012;12:486-490
28. Corr M. Wnt signaling in ankylosing spondylitis. Clinical rheumatology. 2014;33:759-762
29. Sherlock JP, Joyce-Shaikh B, Turner SP. et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. 2012;18:1069-1076
30. Miao CG, Yang YY, He X. et al. Wnt signaling pathway in rheumatoid arthritis, with special emphasis on the different roles in synovial inflammation and bone remodeling. Cell Signal. 2013;25:2069-2078
31. Zhou S, Matsuyoshi N, Liang SB. et al. Expression of T-cadherin in Basal keratinocytes of skin. J Invest Dermatol. 2002;118:1080-1084
Author contact
![]() Corresponding author: Hongzhong Jin, Department of Dermatology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1, Shuai Fu Yuan Hu Tong, Beijing, China 100730. E-mail:jinhongzhongcn
Corresponding author: Hongzhong Jin, Department of Dermatology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1, Shuai Fu Yuan Hu Tong, Beijing, China 100730. E-mail:jinhongzhongcn