Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and Method
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(9):840-852. doi:10.7150/ijms.19268 This issue Cite
Research Paper
The Simultaneous Inhibitory Effect of Niclosamide on RANKL-Induced Osteoclast Formation and Osteoblast Differentiation
Fei-Lan Liu1,2#, Chun-Liang Chen3#, Chia-Chung Lee3, Cheng-Chi Wu1,3, Teng-Hsu Hsu1, Chang-Youh Tsai1, Hsu-Shan Huang3, Deh-Ming Chang1,2,3 ![]()
1. Rheumatology/Immunology/Allergy, Taipei Veterans General Hospital, Taiwan, Republic of China
2. Graduate Institute of Medical Sciences, National Defense Medical Center, Taiwan, Republic of China
3. Graduate Institutes of Life Sciences, National Defense Medical Center, Taiwan, Republic of China
# Both authors contributed equally to this work.
Received 2017-1-20; Accepted 2017-4-23; Published 2017-7-19
Abstract
The bone destruction disease including osteoporosis and rheumatoid arthritis are caused by the imbalance between osteoblastogenesis and osteoclastogenesis. Inhibition of the NF-κB pathway was responsible for decreased osteoclastogenesis. Recently many studies indicated that niclosamide, the FDA approved an antihelminth drug, inhibits prostate and breast cancer cells growth by targeting NF-κB signaling pathways. This study evaluated the effects of niclosamide on osteoclast and osteoblast differentiation and function in vitro.
In RANKL-induced murine osteoclast precursor cell RAW264.7 and M-CSF/RANKL-stimulated primary murine bone marrow-derived macrophages (BMM), niclosamide dose-dependently inhibited the formation of TRAP-positive multinucleated osteoclasts and resorption pits formation between 0.5uM and 1uM. In addition, niclosamide suppressed the expression of nuclear factor of activated T cells c1 (NFATc1) and osteoclast differentiated-related genes in M-CSF/ RANKL-stimulated BMM by interference with TRAF-6, Erk1/2, JNK and NF-κB activation pathways. However, the cytotoxic effects of niclosamide obviously appeared at the effective concentrations for inhibiting osteoclastogenesis (0.5-1uM) with increase of apoptosis through caspase-3 activation in osteoblast precursor cell line, MC3T3-E1. Niclosamide also inhibited ALP activity, bone mineralization and osteoblast differentiation-related genes expression in MC3T3-E1. Therefore, our findings suggest the new standpoint that niclosamide's effects on bones must be considered before applying it in any therapeutic treatment.
Keywords: Niclosamide, Osteoclast Formation, Osteoblast Differentiation
Introduction
Bone is a dynamic tissue consisting of various types of cells which are undergoing renewal and repair process termed “bone remodeling”. The osteoclasts and osteoblasts are major cells types for bone remodeling [1]. The increase in number of osteoclasts could contribute to extreme bone resorption and the decrease in differentiation of osteoblast to reduce new bone formation, disrupts the balance bone remodeling, and results in the loss of bone that are pathological hallmarks of osteoporosis, inflammatory joint disease and rheumatoid arthritis [2-5].
Osteoblasts are derived from mesenchymal stem cells (MSCs) and located in the bone remodeling unit. The formation of bone involves a complex series of process steps which include the proliferation and differentiation of osteoprogenitor cells giving rise to formation of alkaline phosphatase (ALP), bone matrix proteins such as type I collagen and diverse non-collagenous proteins, including osteopontin. and osteocalcin [6]. Osteoblasts differentiation is controlled by a key transcription factor, runt-related transcription factor 2 (Runx2) [7]. Some studies show that Runx2 knockout mice exhibited loss of bone formation and died at birth [8-10]. Runx2 is considered as an essential regulator of osteogenesis and bone formation.
Multinucleated osteoclasts originate from the monocyte/macrophage hematopoietic lineage progenitors that resorb bone in the presence of macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL) [11]. M-CSF is produced by immune cells and osteoblasts that stimulates RANK expression in osteoclast precursor cells and provides a survival signal for osteoclasts [12]. RANKL is secreted by osteoblast and activated T cells that is the main factor involved in the osteoclast differentiation. Binding of RANKL to its receptor RANK activates tumor necrosis factor receptor-associated factor 6 (TRAF6) and subsequently the activation of Jun N-terminal kinase (JNK), extracellular signal regulated kinase (ERK), p38, nuclear factor kappa B (NF-kB) and nuclear factor of activated T-cell (NFAT) c1 which are all the RANK activates key signaling pathways in osteoclasts [13-17].
Salicylanilides were found to be inhibitors of TNF-a via inhibition of NF-kB activation to show anti-inflammatory effects [18]. Derivatives of salicylanilides have multiple pharmacological effects, including anti-bacterial, anti-fungal and anti-cancer and anti-inflammatory activity [19-23]. Accordingly, our previously study have demonstrated NDMC101, a salicylanidide derivative, inhibited RANKL-induced osteoclast differentiation by suppressing NFATc1 and NF-KB activity, and ameliorates paw swelling in collagen-induced arthritis mice [24]. In addition, a new salicylanilide derivatives 6d and 6i, that was synthesized from the lead compound NDMC101, could also suppressed RANKL-stimulated osteoclastogenesis [25]. On the basis of these findings, salicylanilide-derived small molecules could have potential as anti-osteoclastogenesis drugs. Niclosamide, a FDA approved anthelmintics drug, is an analog of salicylanilide and has lately been shown to possess anticancer and atni-inflammatory activities via regulating multiple cellular pathways including the NF-kB, STAT3 and Wnt/β-catenin signaling pathways [26-28]. Recently, cheon et al. reported that niclosamide could suppress osteoclast differentiation [29]. However, the exact signaling mechanism and function of niclosamide on bone remodeling, osteoclast and osteoblast differentiation, are not clear. In the present study, we investigated niclosamide on osteoclast differentiation in RANKL-induced RAW264.7 and BMMs and on osteoblast differentiation in MC3T3-E1 cells, and explore the possible molecular mechanisms.
Material and Method
Mice and reagents
DBA/1J mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All animal experimental procedures were approved by the Animal Care and Use Committee of the National Defense Medical Center, Taiwan (IACUC:15-303). The murine monocyte RAW 264.7 cell line was purchased from the Food Industry Research and Development Institute, Taiwan. MC3T3-E1 was purchased from ATCC, USA. Fetal bovine serum, a-minimum essential medium (a-MEM), penicillin and streptomycin were purchased from Gibco (Grand Island, NY, USA) RANKL and M-CSF were purchased from Peprotech (London, UK). Antibody against TRAF-6 was obtained from Abcam (Cambridge, MA, USA) Anti-JNK, annti-ERK, anti-p38, anti-NF-κB p65, anti- IκBα, anti-phospho-ERK, anti-phospho-JNK, anti-phospho-p38, Anti-pospho-NF-κB p65, anti-phospho-IκB and anti-caspase-3 antibodies were all obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-NFATc1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Niclosamide, anti-ß-actin and anti-lamin B1 antibodies and all other reagents were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Bone marrow macrophage (BMM) isolation
We isolated bone marrow macrophages from mice for osteoclast progenitor cells preparation as described previously [30]. Briefly, Bone marrow cells were isolated from femurs of 8- to 10-week-old DBA/1J mice by flushing the bone marrow cavity with PBS. Cells were incubated with a-MEM containing with 10% heat-inactivated FBS overnight to separate the floating cells. The floating cells were collected and incubated in a-MEM with 10% FBS containing 30ng/ml M-CSF for 3 days to form macrophage-like osteoclast precursor cells.
Cell viability assay
Cell were seeded in a 96 well plate with medium supplemented with 10% fetal bovine serum (FBS) and treated with differentiation concentrations of niclosamide for indicated time, and then treated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (medium containing 0.5mg/ml MTT) for 2 hours at 37˚C. Cells were solubilized in 100 μl DMSO. The intracellular purple formazan concentrations were determined at 550 nm in an ELISA plate reader. The effects of the concentration of niclosamide on cell viability were shown as relative activity (% of concentration of niclosamide-treated group relative to the DMSO control group)
Osteoclast differentiation
We induced osteoclast differentiation as described previously [30]. Briefly, the murine monocyte/macrophage cell line RAW264.7 or macrophage-like osteoclast precursor cells (BMM) were suspended in a-MEM medium containing 10% FBS in the presence of 30 ng/ml M-CSF and 100 ng/ml RANKL or 100 ng/ml RANKL only respectively, as well as differentiation concentrations of niclosamide for 5 days to generate osteoclasts. On Day 3, the medium was replaced with fresh medium containing M-CSF, RANKL, and niclosamide.
Tartrate-resistant acid phosphatase (TRAP) staining
Cells were washed with PBS and fixed with the fixing solution (65:25:8 acetone/citrate solution/37% formaldehyde) for 5 min. After washing with PBS, cells were incubated in the reaction mixture of the Leukocyte Acid Phosphatase Assay kit (Cat. 387, Sigma) at 37˚C for 1 h, away from light, as directed by the manufacturer. Cells were washed 3 times with distilled water and TRAP-positive multinucleated cells containing 5 or more nuclei were visualized by light microscopy and counted as mature osteoclasts.
Bone resorption assay
To assess the effect of niclosamide on RANKL-induced bone resorption, we performed a pit formation assay as described previously [31]. The BMMs or RAW264.7 were seeded into a 24-well Corning Osteo assay plate. All cultures were incubated in triplicate, and cells were replenished every 3 days with fresh osteoclasteogenesis medium with or without different concentration of niclosamide. After 5 days, the wells of plate were treated with 1N NH4OH for 5 min to remove the attached cells. The ratio of the resorbed area to the total area were determined under a microscope in optical fields and quantified using the image J software.
Total RNA extraction and quantitative PCR (Q-PCR)
Total cellular RNA was extracted from cells with different treatment by using the NucleoSpin RNAII kit (Macherey-Nagel). For quantitative RT-PCR analysis, 1 μg of total RNA was converted into cDNA using oligo (dT) and reverse transcriptase (Applied Biosystems). Quantitative PCR was performed on an ABI-Prism 7500 PCR cycler (ABI) using SYBR Green qPCR master mix (Thermo). The PCR primers were listed in supplementary materials. β-actin was used as the house-keeping gene in all PCR analyses, and the ∆∆Ct method was used for quantification.
Western blot analysis
Total proteins were isolated from the cell extracts by using the protein extraction reagent RIAP (Millipore). The equal amounts of proteins were separated on the 12% SDS-PAGE gels and transferred onto PVDF membranes (Amersham). The blots were stained with individual antibodies and followed by a secondary staining with peroxidase-conjugated anti-goat IgG (Sigma), anti-mouse IgG(Jackson) or anti-rabbit IgG (Jackson). The protein bands were visualized using an ECL system (Amersham) and exposing a clear blue X-ray film (Thermo Fisher Scientific) to the membrane.
NFATc1 immunofluorescent staining
To assess the effect of niclosamide on RANKL-induced NFATc1 translaction, we performed a immunofluorescent staining as described previously [31]. BMM were seeded onto glass coverslips and then incubated with 30 ng/ml M-CSF and 100 ng/ml RANKL in the presence or absence of Niclosamide(0.5 or 1uM). After 24 h stimulation, Coverslips were removed, washed in PBS, fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, incubated with 5% bovine serum albumin, and incubated overnight with a specific anti-NFATc1 monoclonal antibody (1:50). Cells were washed in PBS, and then incubated 2 h with FITC-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). After the immunostaining procedures, cells were nuclear-stained with DAPI (Thermo Fisher Scientific). Fluorescence was visualized using a Leica fluorescence microscope.
Flow cytometric Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) assay
We used an Annexin V/7-AAD apoptosis kit (BD Biosciences, San Jose, CA, USA) to assess apoptosis according to the manufacturer. Briefly, the mouse calvaria-origin cell line MC3T3-E1 (ATCC, USA) seeded in 24-well plates in osteoblast differentiation medium with or without various concentration of niclosamide (0, 0.25, 0.5 and 1µM) for 4 days and collected by trypsinization. Cells were resuspended in 400 µl ice-cold 1X binding buffer at a density of nearly 1×106 cells/ml, and then incubated with 10 µl Annexin V-PE/7-AAD for 10 min at room temperature in the dark. The AnnexinV-PE and 7-ADD-labelled cells were analyzed by a flow cytometer. The data was analyzed using WinMDI software.
Osteoblast Differentiation of MC3T3-E1 Cells
MC3T3-E1 (purchased from ATCC, USA) was maintained in α-MEM (Gibco BRL) with 10% heat-inactivated FBS. The cells were grown at 37 °C in a humid atmosphere containing 5% CO2. For the determination of osteoblast differentiation, the cells were seeded a in a 24-well plate and cultured in osteoblast differentiation medium (α-MEM containing 10% FBS, 50 μg/ml ascorbic acid (Sigma-Aldrich) and various concentrations of niclosamide for 7 or 21days. Cell culture media were changed every 3-4 days. After 7 days and 21 days, ALP staining or ALP activity and ECM calcification of the osteoblastic MC3T3-E1 cells were measured respectively.
ALP Staining and ALP activity
ALP staining of the osteoblastic MC3T3-E1 cells was performed using the ALP staining kit (Sigma-Aldrich 86R-1KT). After osteoblast differentiation, cells were fixed with the fixing solution (65:25:8 acetone/citrate solution/37% formaldehyde) for 10 min. The ALP staining of cells was assayed using the enzymatic kit according to the manufacturer's instructions. The stained cells were washed extensively with distilled water and imaged using a camera.
ALP activity of the osteoblastic MC3T3-E1 cells was performed using the ALP reagent containing p-nitrophenyl phosphate (p-NPP, Sigma; N7650) as the substrate. Briefly, after osteoblast differentiation, the cell monolayer was lysed with radioimmunoprecipitation assay buffer (RIPA; Millipore). The clear cell lysates were used for the measurement of ALP activity. The absorbance of p-nitrophenol formed was measured by ELISA reader at a wavelength of 405 nm. The total protein content was determined by a bicinchoninic acid protein assay kit (Pierce, Thermo Scientific, America) and the ALP activity OD values was normalized to the total protein content.
Measurement of ECM Calcification
The Alizarin red S staining method was used to determine the ECM calcification of osteoblasts in vitro. After culture protocol as described above, cells were fixed with 4% paraformaldehyde for 30 min and washed twice with distilled water prior to staining. Then, cells were stained with 40 mM Alizarin red S (Sigma-Aldrich) for 10 min, after which the cells were washed three times with distilled water. Alizarin red S staining for calcium precipitation was imaged using a camera. To quantify the ECM calcium deposits in the cell matrix, the staining of cells was eluted with 100 μL of 10% cetylpyridinium chloride and quantified by measuring the absorbance at 540 nm. The absorbance was shown as relative activity (% of the compound-treated group relative to the DMSO control group).
Statistical analyses
The data were expressed as the means± SD. Statistical analyses were performed by one-way ANOVA followed by Tukey post-hoc test. Values of P<0.05 were considered statistically significant.
Results
Effect of niclosamide on cell viability of osteoclast precursor cell line RAW264.7 and primary murine BMM growth
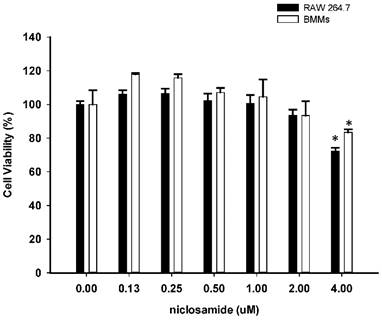
To determine whether niclosamide influences cell viability of RAW264.7 and bone marrow-derived macrophages (BMMs), both of cells were treated with niclosamide (0, 0.13, 0.25, 0.5, 1, 2 and 4 uM) for 24 hours, and cell viability was examined by MTT assay. Results from MTT assay showed that there was no significant change in cell viability after treatment with niclosamide at concentrations ranging from 0.13 to 2 uM. At a concentration of niclosamide 4uM, cell viability of RAW264.7 cells and BMM were approximately 72% and 81% respectively (Fig 1.).
Effect of Niclosamide on cell survival percentage in RAW 264.7 cells and murine bone marrow-derived macrophages (BMM) based on MTT assay. BMM cells were seeded on 96 well plate. Cells were incubated with the indicated concentration of niclosamide for 24 h. After 24h, MTT assay was performed. Cell viability was determined relative to the DMSO control. Data were represented as the means ± SD (n=3). *P<0.05, compared with the 0 uM (DMSO) group.
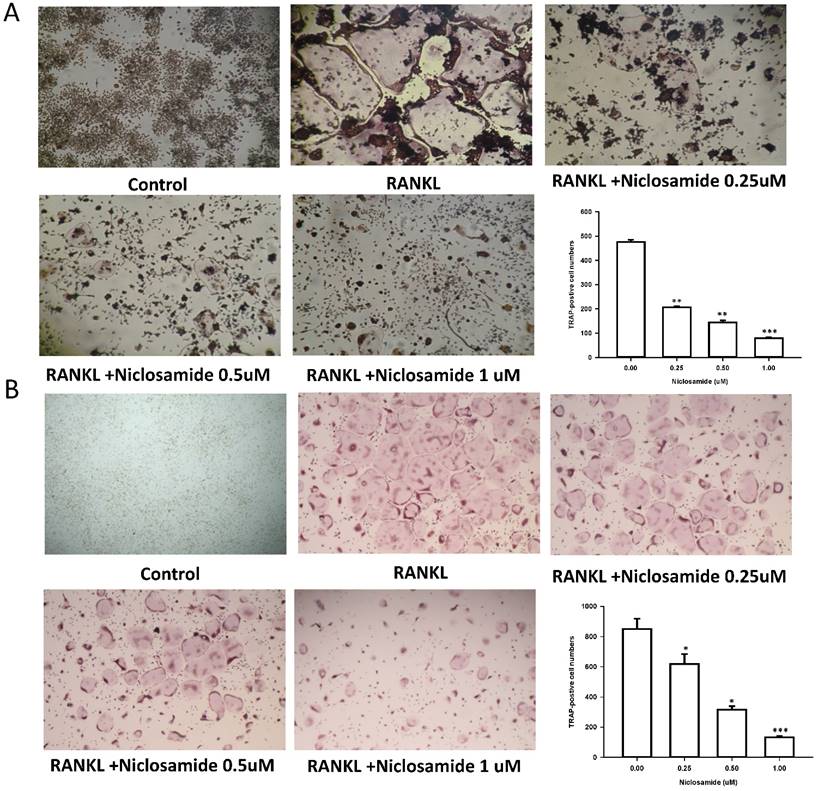
Effect of niclosamide on inhibits RANKL-induced osteoclast differentiation
In order to determine whether niclosamide affects RANKL-induced osteoclast differentiation, RAW 264.7 were culture in the presence of RANKL with or without differentiation concentration of niclosamide for 5 days, then evaluated the inhibitory effects of niclosamide by using TRAP-staining activity assay. Fig 2A showed that niclosamide decrease the number on TRAP positive multinucleated osteoclasts in a dose-dependent manner. The effect of niclosamide on osteoclastogenesis was further confirmed by using primary murine BMM. BMM were form from bone marrow cells treating with M-CSF (30 ng/ml) for 3 days. Then, BMM were stimulated with M-CSF and RANKL together with or without different concentration of niclosamide for 5 days. BMM cultured in the presence of RANKL formed multinucleated TRAP-positive multinucleated osteoclasts. In addition, niclosamide showed inhibition of RANKL-induced osteoclast formation in a concentration dependent manner (Fig 2B). The IC50 value of niclosamide for TRAP-positive multinucleated osteoclast formation from RAW264.7 and BMM were about 0.25 and 0.5 uM, respectively. Niclosamide did not affect the both cells viability when used at concentration that significantly inhibited TRAP-positive osteoclast formation.
Effect of Niclosamide on RANK-induced osteoclast differentiation of RAW264.7 and on MCSF- and RANKL induced osteoclast differentiation of BMM. (A). RAW 264.7 cells were treated with or differentiation concentration of Niclosamide in the presence of RANKL(100ng/ml) for 5 days. (B) BMM treated with or without niclosamide in present MCSF(30ng/ml) and RANKL(100ng/ml) for 5days. After the culture, the cells were fixed and stained by TRAP. The TRAP-positive multinucleated cells containing more than 5 nuclei were considered as mature osteoclast. The data represent mean ± SD (n=3). *P<0.05, compared with the 0 uM (DMSO) group.
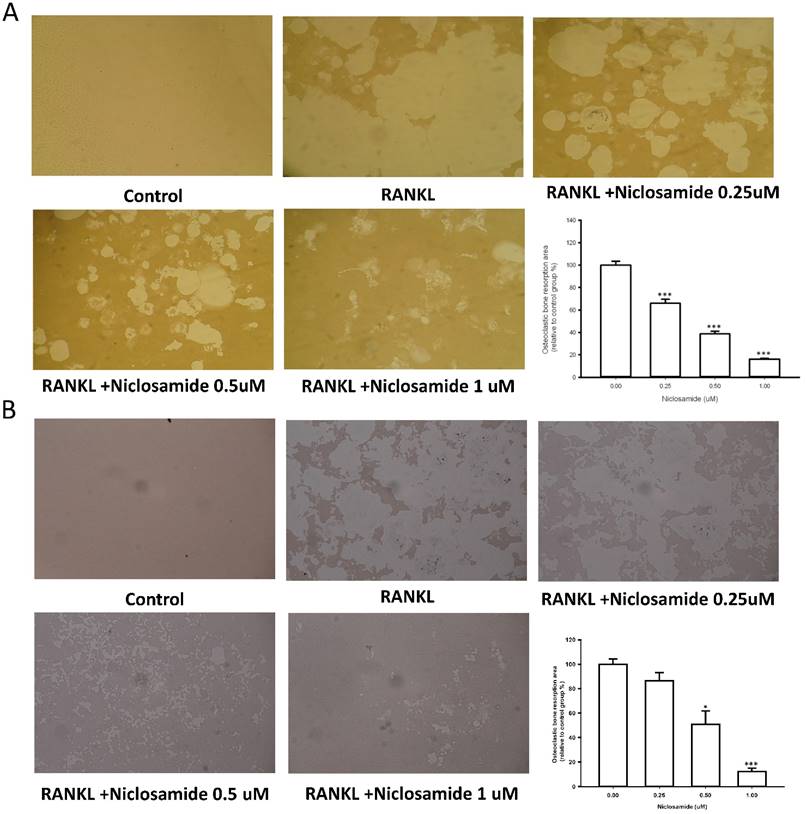
Effect of niclosamide on bone resorption activity in cellture
We used resorption pit formation assay to examine whether niclosamide inhibited osteoclastic bone resorption. We seeded RAW264.7 or BMM onto dentine slices with or without niclosamide in presence of RNAKL for 5 days, and then quantified the formation of resorption pit on the bone slices, in order to determine the direct effects of niclosamide on bone resorption activity. RANKL-induced bone resorption was diminished by niclosamide by dose dependent manners in both RAW264.7 and BMM (Fig 3A and 3B).
Effects of niclosamide on resorption pit formation. BMMs were cultured on bone slices with differentiation concentrations of niclosamide in the presence of MCSF(30ng/ml) and RANKL(100ng/ml) for 5days. After incubation, resorbed lacunae on the bone slices were visualized using light microscopy. The percentages of resorbed area relative to control group were calculated using ImageJ software. *, p < 0.05 and **, p < 0.01 compared with RANKL-treated group.
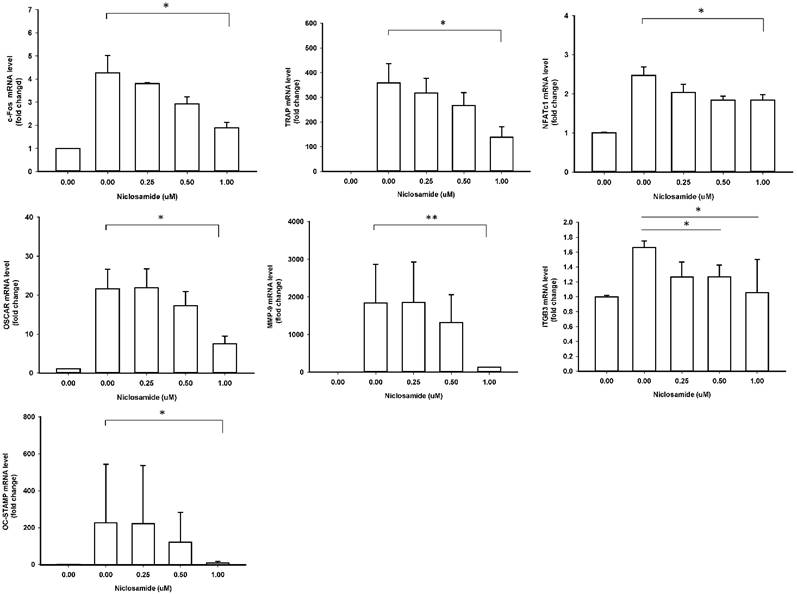
Effect of Niclosamide on RANKL-induced osteoclast specific gene expression in BMM cultures
In order to identify the inhibitory effects of niclosamide on osteoclastogenesis, we examined the RANKL-induced osteoclast specific gene expression in BMM by real-time PCR, including c-fos, NFATC1, TRAP, Cathepsin K, MMP-9, DC-STAMP and ß3 integrin. In mouse BMMs treated with RANKL for 48 h mRNA expressions of all 7 genes were upregulated in the control group, respectively; these genes were significantly inhibited by treating with 1uM niclosamide (Fig 4). The results indicated that niclosamide might suppress RANKL-induced osteoclastgenesis via downregulating the mRNA expression of transcription factors, c-fos and NFATC1.
Effect of Niclosamide on RANKL-induced osteoclast specific gene expression in BMM cultures. Murine bone marrow-derived macrophages (BMM) were pretreated with or without niclosamide for 1 h in medium containing MCSF (30ng/ml) and with RANKL (100 ng/ml) for another 48 hours. Total RNA was isolated with TRIzol, and 1 μg of total RNA was used to synthesize cDNA. cDNA was used as a template for real-time PCR with mouse-specific primers. The relative expression of each target gene compared to β-actin was calculated using the 2-ΔΔCt. Data represent means ± SD of more 3 cultures. *p < 0.05, **p < 0.01, significantly different from values after treatment with DMSO and RANKL.
Effect of niclosamide on RANKL-induced TRAF6 expression
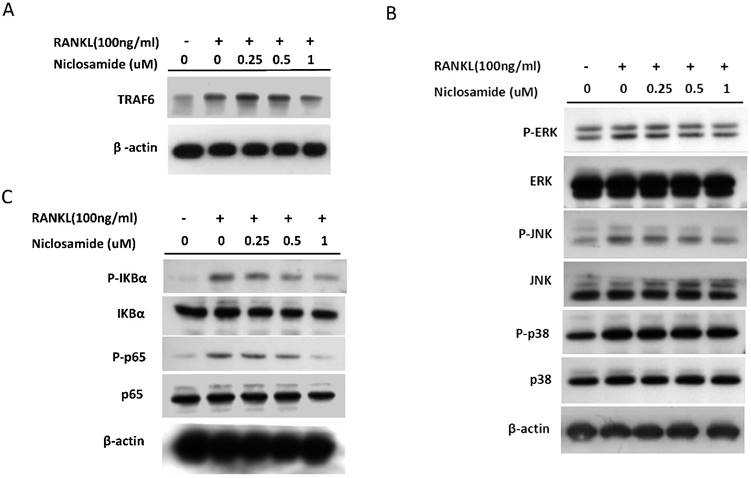
TRAF6 is the primary signaling adaptor in RANK/RANKL signaling and activation of TRAF6 is an essential in initiating ostoeclastgenesis [32]. Because niclosamide inhibited osteoclast differentiation, we determined whether niclosamide inhibited osteoclatogenesis through suppression of TRAF6 protein expression. Fig 5A showed that RANKL induced TRAF6 protein expression in BMM after 24 hours treatment. In addition, pre-treatment with niclosamide markedly suppressed RANKL-induced TRAF6 protein expression in a dose-dependentent manner.
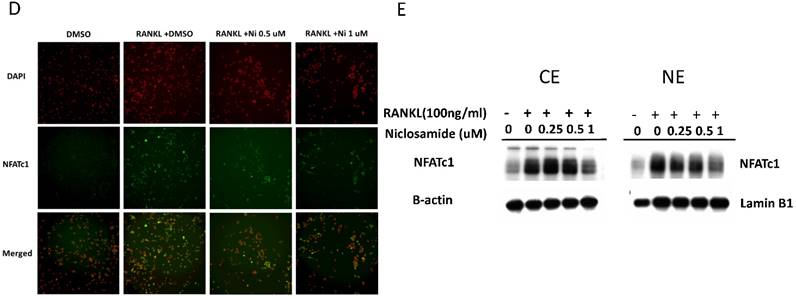
Effect of niclosamide on RANKL-induced TRAF-6 expression, MAPK and IkB/NF-kB activtaion and nuclear translocation of NFATc1 in BMM. (A) Murine bone marrow-derived macrophages (BMM) were pretreated with or without niclosamide for 1 h in medium containing MCSF (30ng/ml) and with RANKL (100 ng/ml) for another 24 hours. The cell extracts were subjected to western blotting assay. (B-C) Murine bone marrow-derived macrophages (BMM) were pretreated with or without niclosamide for 1 h in medium containing MCSF (30ng/ml) and with RANKL (100 ng/ml) for another 30 min. The cell extracts were subjected to western blotting assay. (D) Immunofluorescent analysis. BMM were cultured with or without RANKL (100 ng/ml) in the presence of M-CSF (30 ng/ml). Niclosamide was added to some of the cultures treated with RANKL. After 24 h, the cells were stained with anti-NFATc1 antibody (green) and DAPI (nuclear staining, red) (upper panels). The nuclear localization of NFATc1 was confirmed in merged images of BMM treated with RANKL (yellow).
Effect of niclosamide on RANKL-induced signaling pathway in BMM cultures
The report showed that TRAF6 plays a critical role in RANKL-induced osteoclast differentiation via activation of phosphorylation of MAPKs (ERK, JNK and p38) in osteoclast precursor cells [33]. Based on the findings that niclosamide suppressed RANKL-induced TRAF6 protein expression (Fig 5A), we next investigated the effect of niclosamide on RANKL-induced ERK, JNK and p38 phosphrylation in BMM by western blotting. RANKL activated ERK, JNK and p38 phosphorylation at the time point of 30 min. Fig. 5B indicated that niclosamide inhibited RNAKL-induced phosphorylation of ERK and JNK in a dose-dependent manner, but niclosamide did not influence RNAKL-induced p38 phosphorylation.
The nuclear factor kappa B (NF-κB) is also the downstream target of TRAF6 transmits the RANKL/RANK signal. Activation of NF-kB plays a dominant role in initiating osteoclastogenesis [34]. We used western blot assay to investigate whether niclosamide inhibited osteoclast formation via suppression of NF-kB signaling pathway in BMM. Fig. 5C showed that pretreatment of niclosamide for 2 hours could suppress RANKL-induced phosphorylation of IkBa and NF-kB p65 in BMM. Together our result indicated that niclosamide might abrogate osteoclast differentiation and bone resorption via the inhibition of RANKL-induced ERK, JNK MAPKs and NK-kB activation.
Effect of niclosamide on RANKL-induced nuclear translocation and expression NFATc1
The several transcription factors which promote osteoclastic gene expression are induced by RANKL and are considered to act downstream of the RANKL signaling pathway [35]. The activation of early-RANKL-response factor, such as NF-kB, is activated before late-RANKL-response factor, NFATC1, a key transcription factor involved in osteoclast differentiation [36]. The inactive forms of NFATc1 are localized in the cytoplasm in the cells. However, NFATC1 translocation from cytoplasm to nucleus will have an ability to activate gene transcription [37]. As a result, we investigated the effect of niclosamide on the nuclear translocation of NFATc1 in RANKL-stimulated BMM by immunofluorescence staining. As shown in Fig 5D, nuclear accumulation of NFATC1 was increased by RANKL stimulation for 24 hours compared with the level in BMM without RANKL treatment. Next, we performed western blot assay to further confirm whether niclosamide inhibited nfatc1 nuclear translocation at the protein level. Figure 5E indicated that NFATc1 in cytoplasmic and nuclear extracts in BMM were increased after 24 treatments by RANKL. Treatment of niclosamide inhibited NFATc1 expression in cytoplasm and NFATc1 translocation to nuclear in BMM. These results indicated that niclosamide negatively regulated RANKL-induced NFATc1 expression and nuclear translocation during osteoclastogenesis in BMM.
Effect of niclosamide on the cell viability of Mc3T3-E1 cells
Bone homeostasis is maintained by a balance of osteoclast and osteoblast. We test the effect of niclosamide on the cell viability of the Mc3T3-E1 by MTT assay. As shown in Fig 6A, niclosamide has significant inhibition on Mc3T3-E1 viability at the concentrations of 0.5uM to 1uM after 1 day treatment, which were enhanced after 2 and 3 days.
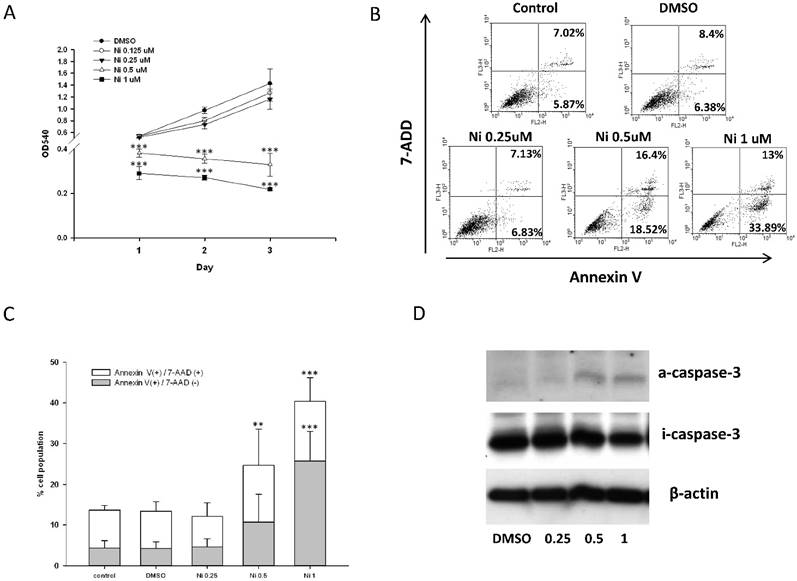
Effect of niclosamide on the cell viability, apoptosis and caspase-3 activation of MC3T3-E1 cells. (A) The MC3T3-E1 cells were seeded onto 96 well and incubated with osteoblast differentiation medium containing with the indicated concentration of niclosamide for 1, 2 and 3 days. MTT assay was performed. Data are represented as mean ± SD (n = 3) and were subjected to statistical analysis, ***P < 0.001 versus DMSO control. (B) The MC3T3-E1 cells were incubated with osteoblast differentiation medium containing with the indicated concentration of niclosamide for double staining with annexin-V-PE and7-AAD, followed by flow cytometry analysis (C) The percentage of annexin-V-PE(+) /7-AAD(-) and annexin-V-PE(+) /7-AAD(+) cells in each group, presented as graphs. Data are represented as mean ± SD (n = 3) and were subjected to statistical analysis, ** P < 0.01 and ***P < 0.001, versus the DMSO control. (C) The protein level of activity (a) and inactivity (i) caspase-3 were detected by western blot and B-actin as internal control.
Effect of niclosamide on apoptosis of MC3T3-E1 cells
To determine whether the inhibition of MC3T3-E1 cells viability following niclosmaide was because of apoptosis, we performed Annexin V/ 7 AAD flow cytometric analyses to distinguish among healthy cells (Annexin V negative and 7 AAD negative), early apoptotic cells (Annexin V positive and 7 AAD negative) and late apoptotic cells. MC3T3-E1 cells incubated with 0.25 uM niclosamide for 4 days, the percentage of the three types cell populations were not significantly different to control groups (0uM niclosamide and DMSO treatment). On the other hand, treatment of MC3T3-E1 cells with niclosamide (0.5 and 1uM) for 4 days provoked a dose-dependent increase in the number of early apoptotic cells and late apoptotic cells (Fig 6B and 6C).
The activation of caspase-3 which is a protease involved in the initiation of the apoptosis pathway, eventually causes cell apoptosis [38]. In order to analyze whether niclosamide induced MC3T3-E1 apoptosis through activation of caspase-3, we collected cells total protein from MC3T3-E1 stimulated with or without niclosamide (0.25, 0.5 and 1uM) for 2 days for western blot assay. As shown in Fig 6D, the protein level of inactive caspase-3 was upregulated at niclosamide concentrations of 0.5 and 1 uM. Together, these results indicated that the inhibition of MC3T3-E1 cell viability by niclosamide was caused by the activation of caspase-3 and apoptosis.
Effect of niclosmide on the differentiation of MC3T3-E1
As the expression of ALP is a phenotypic marker for the early osteoblast differentiation, the effect of niclosamide on ALP activity was examined using the alkaline phosphatase activity kit and ALP staining (Fig 7A). After 7 days of niclosamide treatment, histochemicalanalysis of the ALP stain showed that niclosamide caused a dose-dependent decrease on ALP activity in Mc3T3-E1. In addition, niclosamide decreased the ALP activity of the MC3T3-E1 cells in a dose-dependent manner after 7 days exposure (Fig 7B).
Effect of Niclosamide on osteoblast differentiation of MC3T3-E1 cells. Cells were treated with the different concentration of niclosamide in the presence Osteoblast differentiation medium for 7 or 21 days, and then the effects of the niclosamide on ALP activity and ECM calcification were evaluated by the ALP or Alizarin red S staining methods. (A) ALP staining on day 7. (B) Alizaring red S staining on day 21. (C) ALP activity on day 7. (D) ECM calcification was quantified by the amount of cell-bound Alizarin red S. Alizarin red S was released by incubation in 10% cetylpyridinium chloride and then detected by measuring the absorbance at 540 nm. ECM calcification was determined relative to the DMSO control. Data are presented as the mean ± SD of three independent experiments. (E) Gene expression levels of RUNX-2, ALP, Col I and OCN on day 7 were determined by real-time PCR analysis. The data were normalized to GPADH. * P < 0.01 and ***P < 0.001, versus the DMSO control.
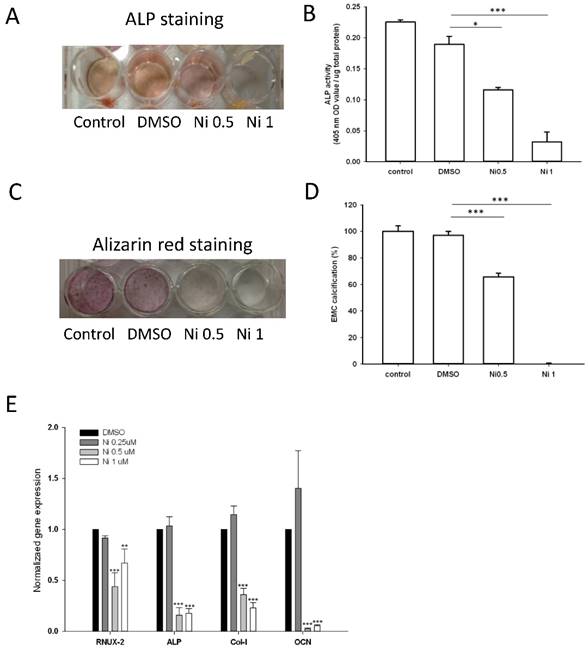
The formation of mineralization nodules is a marker of osteoblastic maturation [39]. We examined the effect of niclosamide on osteoblast maturation by using alizarin red S-positive staining. Figure 7C showed that MC3T3-E1 exhibited alizarin red S positive staining after treated with osteoblast differentiation medium for 21 days. In contract, the formation of mineralization was significantly reduced by niclosamide in a dose dependent manner (Fig 7C). Quantitatively, niclosamide significantly reduced alizarin red S staining in MC3T3-E1 under osteoblast differentiation medium. The 1 uM niclosamide treatment, the extracted mineralized stain from Mc3T3-E1 dropped by approximately 98-100% (Fig 7D).
We further examined the effect of niclosamide on the expression level of the critical genes involved in osteogenic differentiation, RUNX2, COL I, ALP and OCN by quantitative RT-PCR. The results of quantitative RT-PCR analysis showed that the cell treated with niclosamide for 7 days exhibited downregulation in the expression level of all genes. These data suggest that niclosamide at the concentrations >0.5 uM suppressed MC3T3-E1 cells differentiation and maturation (Fig 7D).
Discussion
Osteoporosis is attributed to an imbalance between bone resorption and bone formation with a net bone loss. The many clinical drugs are currently used in osteoporosis treatment, which are broadly categorized as either anabolic or anti-resorptive. Such as, bisphosphonates, estrogen, and calcitonin are all bone resorption inhibitors, which maintain bone mass by inhibiting the function of osteoclasts. However, most of these drugs are associated with some side effects, including thromboembolism, hypercalcemia and breast cancer [40-42]. Since osteoclasts are responsible for bone resorption and they are one of the main targets for treatment of osteoporosis, therefore developing new drugs for therapies that have both anabolic and anti-resorption effects would be more effective in treating bone loss in osteoporosis. However, drug discovery and development is a complex, lengthy and costly procedure [43]. Finding new uses for existing drugs is a proven short cut between the lab and the clinic, which can save production cost and time [44].
In the current study, niclosamide a teniacide in the anthelmintic family has been indicated as a potential anticancer agent to offer a new approach on treating cancer. Niclosamide can block the multiple signaling pathways, including Wnt/β-catenin pathway, mTORC1 pathway, STAT3 and NF-kB pathways to have anti-cancer effect on human breast cancer [45], lung cancer cells [46], prostate cancer [45], colon cancer [47], and ovarian cancer [48].NF-kB pathway is an essential pathway for RANKL-induced osteoclast differentiation and activation [49]. Together all reports, niclosamide might be a potential drug target for anti-osteoclast differentiation. Our data showed that niclosamide can inhibit RANKL-induced osteoclast differentiation and function in BMM through suppressed the activation of NF-κB by the inhibition of IκB and NF-kB p65 phosphorylation. We considered NF-κB could be an important downstream pathway that mediates the effects of niclosamide on osteoclast differentiation and function.
NF-kB is one of the important downstream pathways in TRAF-6 mediated-osteoclast differentiation. TRAF-6 is a signaling adaptor molecule in osteoclast precurosrs, and binding of RANKL to RANK is initiated following TRAF6 signaling cascade [50, 51].
In addition to NF-κB, TRAF-6 also activates a series of major intracellular signaling transduction pathways including JNK, p38, and ERK MAPK which plays a crucial role in osteoclasogenesis. In the present study, we examined the inhibitory mechanisms of niclosamide on RANKL/RANK signaling. We found that niclosamide inhibited RANKL-stimulated TRAF-6 expression, subsequently suppressed the phosphorylation of JNK and ERK MAPK, but not p38 MAPK. Collectively, our data suggest that niclosomide inhibits osteoclast differentiation and function via suppressing TRAF6 expression and its downstream pathways, including NF-kB and MAPKs.
Many studies have indicated that NF-kB is an upstream transcription factor regulating expression of NFATc1, a vitaly transcription factor involved in osteoclastogenesis. Because NFATc1-deficient embryonic stem cells exhibit a defect in RANKL-induced osteoclastogenesis, NFATc1 is considered to be an important factor in the regulation of osteoclast formation [35, 52]. NFTAC1 autoamplifies and conducts the expression of conducts the expression of osteoclastogenesis-realted genes, such as TRAP [53], OSCAR [37], cathepsin K [54], DC-STAMP and MMP-9. Our results revealed that niclosamide significantly suppressed RANKL-induced NFATc1 expression and nuclear translocation in BMM and concomitantly inhibited the mRNA expression of NFATc1-mediated osteoclastogenic genes including TRAP, OSCAR, CTK, DC-SATMP and MMP9. Taken together, these findings indicated that niclosamide inhibited osteoclastogenesis via down-regulation of RANKL-induced NFATc1 expression and translocating into nucleus.
Our data showed that the effective concentration (>0.5uM) of niclosamide used on anti-osteoclastogenesis would reduce MC3T3-E1 osteoblast-like cells viability and increase cell apoptosis by enhancing caspase-3 activation. In addition, niclosamide suppressed ALP activity, a vital in initiation of osteoblast differentiation and bone mineralization during bone nodules formation [55, 56], and reduced the expression of osteoblast differentiation-related gene, including RUNX2, OCN, ALP and Col-I [57]. When the niclosamide concentration was 1uM, the cells viability of MC3T3-E1 were evidently reduced and the differentiation and mineralization potentiality of MC3T3-E1 were unable to be find. The activation of Wnt/β-catenin signaling pathway has been reported for a major regulator of bone mass formation [58]. Niclosamide is considered as a potent Wnt/β-catenin inhibitor. This might a reason for niclosamide on inhibition of osteoblast differentiation.
According to our finding, niclosamide could inhibit not only osteoclast formation and function but also osteoblast. Some previous studies indicated that clinical medicines such as SSRIs (serotonin-specific reuptake inhibitors) [59]. and bisophophonates (zoledronate and Alendronate) can suppress osteoclast and osteoblast formation [60]. The observed decreases in osteoclast and osteoblast number and function may contribute to a low bone turnover state in vivo, which may contribute to the pathogenesis of bone disorders such as fragility fracture and osteonecrosis of the jaw [61]. Many studies have demonstrated that niclosamide has potent in vitro and in vivo anti-tumor growth activities and suggested niclosamide for new drugs applications for treatment of cancer [62]. However, Li et. al. summarized the earlier studies to indicate that niclosamide inhibited cell proliferation of breast, lung, prostate, colon and ovarian cancer cell lines, and that the IC50 values for most cell lines were around 0.5 to 1 μM [62]. Our data showed that 0.5 to 1uM niclosamide significantly induced osteoblast apoptosis and suppressed osteoblast differentiation and function. The negative side effects of niclosamide on osteoblast activities should be concerned in further clinical use. Our research findings support the new standpoint that niclosamide's effects on bones must be considered before applying it in any therapeutic treatment.
Conclusion
Our results indicated that niclosamide could inhibit osteoclastogenesis in vitro via the reduced the expression of TRAF6, and inhibition of NF-kB and MAPKs (ERK and JNK) activation and subsequently attenuated the NFTAc1 nuclear translocation and osteoclastogenesis related gene expression. However, niclosamide also negatively regulated bone formation by directly suppressing Osteoblastic MC3T3-E1 cell proliferation, cell viability and inducing cell apoptosis and then inhibiting matrix mineralization. Overall, our study might raise an issue that niclosamide would affect bone turnover by both inhibition of osteoclast and osteoblast differentiation and that this is to be taken in consideration of the bone metabolisms effects in chronic administration of niclosamide, in particular.
Supplementary Material
PCR primers.
Acknowledgements
Fei-Lan Liu was supported by the Ministry of Science and Technology, Taiwan, Republic of China (Most 104-2811-B-075 -006 and Most 105-2811-B-075 -002).
This work was supported by the Ministry of Science and Technology, Taiwan, Republic of China (Most 102-2314-B-075 -083 -MY3 and Most 105-2314-B-075 -054 -MY3)
Study conception, design and writing: Fei-Lan Liu, Chun-Liang Chen and Deh-Ming Chang.
Acquisition of data: Fei-Lan Liu, Chun-Liang Chen, Chia-Chung Lee, Cheng-Chi Wu, and Teng-Hsu Hsu
Analysis and interpretation of data: Fei-Lan Liu, Chun-Liang Chen, Chang-Youh Tsai, Hsu-Shan Huang and Deh-Ming Chang
Proof reading the article: Deh-Ming Chang
All authors reviewed the manuscript.
Competing Interests
The authors have declared no conflicts of interest.
References
1. Goltzman D. Discoveries, drugs and skeletal disorders. Nature Reviews Drug Discovery. 2002;1:784-96
2. Karmakar S, Kay J, Gravallese EM. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheumatic Disease Clinics of North America. 2010;36:385-404
3. Post TM, Cremers SC, Kerbusch T, Danhof M. Bone Physiology, Disease and Treatment. Clinical pharmacokinetics. 2010;49:89-118
4. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-42
5. Cochran DL. Inflammation and bone loss in periodontal disease. Journal of periodontology. 2008;79:1569-76
6. Nakashima K, de Crombrugghe B. Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends in genetics: TIG. 2003;19:458-66
7. Weissmann G. et al. Rheumatologists gauge new uses of old drugs. Hospital practice. 1981;16:37 40-A, D passim
8. Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K. et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755-64
9. Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR. et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765-71
10. Franceschi RT, Xiao G. Regulation of the osteoblast-specific transcription factor, Runx2: responsiveness to multiple signal transduction pathways. Journal of cellular biochemistry. 2003;88:446-54
11. Suda T, Takahashi N, Martin TJ. Modulation of osteoclast differentiation. Endocrine reviews. 1992;13:66-80
12. Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nature reviews Cancer. 2014;14:722-35
13. Galibert L, Tometsko ME, Anderson DM, Cosman D, Dougall WC. The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-kappaB, a member of the TNFR superfamily. The Journal of biological chemistry. 1998;273:34120-7
14. Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y. The TRAF family of signal transducers mediates NF-kappaB activation by the TRANCE receptor. The Journal of biological chemistry. 1998;273:28355-9
15. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-42
16. Feng X. RANKing intracellular signaling in osteoclasts. IUBMB life. 2005;57:389-95
17. Teitelbaum SL. RANKing c-Jun in osteoclast development. The Journal of clinical investigation. 2004;114:463-5
18. Wei J, Feng J. Signaling pathways associated with inflammatory bowel disease. Recent patents on inflammation & allergy drug discovery. 2010;4:105-17
19. Brown ME, Fitzner JN, Stevens T, Chin W, Wright CD, Boyce JP. Salicylanilides: selective inhibitors of interleukin-12p40 production. Bioorganic & medicinal chemistry. 2008;16:8760-4
20. De La Fuente R, Sonawane ND, Arumainayagam D, Verkman AS. Small molecules with antimicrobial activity against E. coli and P. aeruginosa identified by high-throughput screening. British journal of pharmacology. 2006;149:551-9
21. Vinsova J, Imramovsky A, Buchta V, Ceckova M, Dolezal M, Staud F. et al. Salicylanilide acetates: synthesis and antibacterial evaluation. Molecules. 2007;12:1-12
22. Otevrel J, Mandelova Z, Pesko M, Guo J, Kralova K, Sersen F. et al. Investigating the spectrum of biological activity of ring-substituted salicylanilides and carbamoylphenylcarbamates. Molecules. 2010;15:8122-42
23. Wang B, Wang Z, Ai F, Tang WK, Zhu G. A monofunctional platinum(II)-based anticancer agent from a salicylanilide derivative: Synthesis, antiproliferative activity, and transcription inhibition. Journal of inorganic biochemistry. 2014;142C:118-25
24. Cheng CP, Huang HS, Hsu YC, Sheu MJ, Chang DM. A benzamide-linked small molecule NDMC101 inhibits NFATc1 and NF-kappaB activity: a potential osteoclastogenesis inhibitor for experimental arthritis. Journal of clinical immunology. 2012;32:762-77
25. Lee CC, Liu FL, Chen CL, Chen TC, Chang DM, Huang HS. Discovery of 5-(2',4'-difluorophenyl)-salicylanilides as new inhibitors of receptor activator of NF-kappaB ligand (RANKL)-induced osteoclastogenesis. European journal of medicinal chemistry. 2015;98:115-26
26. Ye T, Xiong Y, Yan Y, Xia Y, Song X, Liu L. et al. The anthelmintic drug niclosamide induces apoptosis, impairs metastasis and reduces immunosuppressive cells in breast cancer model. PloS one. 2014;9:e85887
27. Jin Y, Lu Z, Ding K, Li J, Du X, Chen C. et al. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer research. 2010;70:2516-27
28. Osada T, Chen M, Yang XY, Spasojevic I, Vandeusen JB, Hsu D. et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer research. 2011;71:4172-82
29. Cheon YH, Kim JY, Baek JM, Ahn SJ, So HS, Oh J. Niclosamide suppresses RANKL-induced osteoclastogenesis and prevents LPS-induced bone loss. Biochemical and biophysical research communications. 2016;470:343-9
30. Hsu YC, Cheng CP, Chang DM. Plectranthus amboinicus attenuates inflammatory bone erosion in mice with collagen-induced arthritis by downregulation of RANKL-induced NFATc1 expression. The Journal of rheumatology. 2011;38:1844-57
31. Chen CL, Liu FL, Lee CC, Chen TC, Ahmed Ali AA, Sytwu HK. et al. Modified salicylanilide and 3-phenyl-2H-benzo[e][1,3]oxazine-2,4(3H)-dione derivatives as novel inhibitors of osteoclast differentiation and bone resorption. Journal of medicinal chemistry. 2014;57:8072-85
32. Armstrong AP, Tometsko ME, Glaccum M, Sutherland CL, Cosman D, Dougall WC. A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J Biol Chem. 2002;277:44347-56
33. Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S. et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. The EMBO journal. 2001;20:1271-80
34. Bruzzaniti A, Baron R. Molecular regulation of osteoclast activity. Reviews in endocrine & metabolic disorders. 2006;7:123-39
35. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H. et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889-901
36. Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM. et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. The Journal of biological chemistry. 2007;282:18245-53
37. Kim K, Kim JH, Lee J, Jin HM, Lee SH, Fisher DE. et al. Nuclear factor of activated T cells c1 induces osteoclast-associated receptor gene expression during tumor necrosis factor-related activation-induced cytokine-mediated osteoclastogenesis. J Biol Chem. 2005;280:35209-16
38. Ding WX, Ni HM, DiFrancesca D, Stolz DB, Yin XM. Bid-dependent generation of oxygen radicals promotes death receptor activation-induced apoptosis in murine hepatocytes. Hepatology. 2004;40:403-13
39. Niu YB, Li YH, Kong XH, Zhang R, Sun Y, Li Q. et al. The beneficial effect of Radix Dipsaci total saponins on bone metabolism in vitro and in vivo and the possible mechanisms of action. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2012;23:2649-60
40. Lloyd M. Treatment of postmenopausal osteoporosis. The New England journal of medicine. 1998;339:202 author reply 3
41. Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508-14
42. Body JJ. Calcitonin for the long-term prevention and treatment of postmenopausal osteoporosis. Bone. 2002;30:75S-9S
43. Kraljevic S, Stambrook PJ, Pavelic K. Accelerating drug discovery. EMBO reports. 2004;5:837-42
44. Aronson JK. Old drugs-new uses. British journal of clinical pharmacology. 2007;64:563-5
45. Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/beta-catenin pathway. PloS one. 2011;6:e29290
46. Li R, Hu Z, Sun SY, Chen ZG, Owonikoko TK, Sica GL. et al. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non-small cell lung cancer. Molecular cancer therapeutics. 2013;12:2200-12
47. Sack U, Walther W, Scudiero D, Selby M, Kobelt D, Lemm M. et al. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. Journal of the National Cancer Institute. 2011;103:1018-36
48. Yo YT, Lin YW, Wang YC, Balch C, Huang RL, Chan MW. et al. Growth inhibition of ovarian tumor-initiating cells by niclosamide. Molecular cancer therapeutics. 2012;11:1703-12
49. Yu M, Qi X, Moreno JL, Farber DL, Keegan AD. NF-kappaB signaling participates in both RANKL- and IL-4-induced macrophage fusion: receptor cross-talk leads to alterations in NF-kappaB pathways. Journal of immunology. 2011;187:1797-806
50. Poblenz AT, Jacoby JJ, Singh S, Darnay BG. Inhibition of RANKL-mediated osteoclast differentiation by selective TRAF6 decoy peptides. Biochemical and biophysical research communications. 2007;359:510-5
51. Lamothe B, Webster WK, Gopinathan A, Besse A, Campos AD, Darnay BG. TRAF6 ubiquitin ligase is essential for RANKL signaling and osteoclast differentiation. Biochemical and biophysical research communications. 2007;359:1044-9
52. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251-64
53. Shiotani A, Takami M, Itoh K, Shibasaki Y, Sasaki T. Regulation of osteoclast differentiation and function by receptor activator of NFkB ligand and osteoprotegerin. The Anatomical record. 2002;268:137-46
54. Saftig P, Hunziker E, Everts V, Jones S, Boyde A, Wehmeyer O. et al. Functions of cathepsin K in bone resorption. Lessons from cathepsin K deficient mice. Advances in experimental medicine and biology. 2000;477:293-303
55. Wennberg C, Hessle L, Lundberg P, Mauro S, Narisawa S, Lerner UH. et al. Functional characterization of osteoblasts and osteoclasts from alkaline phosphatase knockout mice. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2000;15:1879-88
56. Chaudhary LR, Hofmeister AM, Hruska KA. Differential growth factor control of bone formation through osteoprogenitor differentiation. Bone. 2004;34:402-11
57. Anderson HC. Mechanism of mineral formation in bone. Laboratory investigation; a journal of technical methods and pathology. 1989;60:320-30
58. Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS. et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. The Journal of biological chemistry. 2005;280:33132-40
59. Hodge JM, Wang Y, Berk M, Collier FM, Fernandes TJ, Constable MJ. et al. Selective serotonin reuptake inhibitors inhibit human osteoclast and osteoblast formation and function. Biological psychiatry. 2013;74:32-9
60. Huang X, Huang S, Guo F, Xu F, Cheng P, Ye Y. et al. Dose-dependent inhibitory effects of zoledronic acid on osteoblast viability and function in vitro. Molecular medicine reports. 2016;13:613-22
61. Strampel W, Emkey R, Civitelli R. Safety considerations with bisphosphonates for the treatment of osteoporosis. Drug safety. 2007;30:755-63
62. Li Y, Li PK, Roberts MJ, Arend RC, Samant RS, Buchsbaum DJ. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer letters. 2014;349:8-14
Author contact
![]() Corresponding author: Deh-Ming Chang, MD, PhD., Rheumatology/Immunology/Allergy, Taipei Veterans General Hospital, Taiwan, Republic of China. Tel: +886-2-7735-7799; Fax: +886-2-7735-1333; E-mail: ming0503hinet.net
Corresponding author: Deh-Ming Chang, MD, PhD., Rheumatology/Immunology/Allergy, Taipei Veterans General Hospital, Taiwan, Republic of China. Tel: +886-2-7735-7799; Fax: +886-2-7735-1333; E-mail: ming0503hinet.net