Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Classification of brain injury
Consequences of primary...
Physiological changes observed...
Significance of TBI animal models
Limiting factors in TBI animal...
Recommended Clinical Secondary...
Hyperglycemia
Hyponatremia
Hyperthermia
Hypoxemia/hypotension/ischemia
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(5):494-505. doi:10.7150/ijms.18075 This issue Cite
Review
Improving on Laboratory Traumatic Brain Injury Models to Achieve Better Results
Mark Nyanzu1, 2, Felix Siaw-Debrah1, 2, Haoqi Ni1, 2, Zhu Xu1, 2, Hua Wang1, 2, Xiao Lin1, 2, Qichuan Zhuge1,2 ![]() , Lijie Huang1,2
, Lijie Huang1,2 ![]()
1. Zhejiang Provincial Key Laboratory of Aging and Neurological Disorder Research, First Affiliated Hospital, Wenzhou Medical University, Wenzhou 325000, China;
2. Department of Neurosurgery, the First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325000, China.
Received 2016-10-25; Accepted 2017-1-31; Published 2017-4-9
Abstract
Experimental modeling of traumatic brain injury (TBI) in animals has identified several potential means and interventions that might have beneficial applications for treating traumatic brain injury clinically. Several of these interventions have been applied and tried with humans that are at different phases of testing (completed, prematurely terminated and others in progress). The promising results achieved in the laboratory with animal models have not been replicated with human trails as expected. This review will highlight some insights and significance attained via laboratory animal modeling of TBI as well as factors that require incorporation into the experimental studies that could help in translating results from laboratory to the bedside. Major progress has been made due to laboratory studies; in explaining the mechanisms as well as pathophysiological features of brain damage after TBI. Attempts to intervene in the cascade of events occurring after TBI all rely heavily on the knowledge from basic laboratory investigations. In looking to discover treatment, this review will endeavor to sight and state some central discrepancies between laboratory models and clinical scenarios.
Keywords: traumatic brain injury, secondary insults, animal models
Introduction
Scientist and medical practitioners conduct research on TBI for two main reasons; Firstly, TBI is the number one cause of disability and death in people at the prime of their lives (under 45 years of age) especially in industrialized countries. TBI is often caused by road traffic accidents. Each fatality comes with different levels of brain damage to the survivors ranging from mild, moderate to severe brain damage1. Even though TBI is a major socioeconomic and medical problem, its pathogenesis and mechanism is not fully understood because it is extremely challenging to re-enact the proceedings that led to the primary and secondary brain insults2,3. Comparatively, TBI is quantifiable and performed under controlled environments in the laboratory.
Secondly, research conduction is still ongoing and will continue because although a whole lot of neuro-protective agents have been studied and researched, the very ones that have made or shown promises in laboratory models have failed to provide consistency and strikes in human trials4-7. Evaluating and testing the effectiveness of therapeutic agents in laboratory models will hence continue to be an important precursor to their functions in humans. This review highlights the contributions of different types of animal models especially their strengths in unraveling the neuropathological features of TBI and their weakness that has slowed the translation of experimental results at the bedside. Considerably there has been a lot of investigation into TBI with animal models but no novel therapy has been successfully translated from the bench to the clinic. Despite the fact clinical trials have well described limitations that might be contributing factors to these failures, several modeling limitations account for the lack of therapeutic progress from bench side to the bedside. TBI models usually omit one or more critical and clinically essential pathophysiological feature. In this review, several important clinical pathophysiologic factors in TBI, namely secondary insults (i.e., hypotension and/or hypoxemia), coma, and aspects of standard neuro-intensive care monitoring and management strategies (i.e., intracranial pressure [ICP] monitoring and ICP-directed therapies, sedation, mechanical ventilation, and cardiovascular support) requiring incorporation into animal models are discussed.
Classification of brain injury
Traumatic brain injuries are classified on different levels. In this article we will categorized them from the clinical point of view as either focal or diffuse injuries8. A description concerning brain injures pertains to all kinds of brain injuries in different environments both civilian and military. Both diffused and focal brain injuries have their distinct features at the clinical level, which are usually visible with the help of radiology. Focal brain injuries for example can be easily identified by using standard imaging modalities such as CT and MRI. From table 1, focal brain injury is usually categorized based on the location of the bleeding with respect to the brain: within the brain (intracerebral hematomas; tissue tears), on the surface of the brain (acute subdural hematoma; subarachnoid hemorrhage; extradural hematoma), or in the cortical gray matter (cerebral contusion). All these kinds of focal brain injuries can be observed in severely or moderately injured population. Nevertheless, in mild head injuries the only focal brain injury that appears is subarachnoid hemorrhage. However, because mild brain injuries with subarachnoid hemorrhage are rare, it is appropriate to state that mild brain injuries are predominantly classified as diffuse injuries. Diffuse brain injuries are defined as injuries not located to just one area of the brain but spread or distributed all over the brain. A common component or a typical feature of diffuse brain injuries is brain swelling that appears over time following injury9, 10.
Brain injury classification
| Brain Injuries | |||
|---|---|---|---|
| Primary traumatic brain damage (neural or vascular [or both]) | Diffuse | Diffuse axonal injury (DAI) | |
| Diffuse vascular injury (DVI) | |||
| Focal | Vascular injury resulting in | Intracerebral hemorrhage | |
| Subdural hemorrhage | |||
| Extradural (epidural) hemorrhage | |||
| Axonal injury | |||
| Contusion | |||
| Laceration | |||
| Secondary traumatic brain damage | Diffuse | Diffuse hypoxic-ischemic damage | |
| Diffuse brain swelling | |||
| Focal | Focal hypoxic-ischemic injury | ||
| Focal brain swelling | |||
Consequences of primary traumatic brain injury
Loss of consciousness
Acceleration of human head produces different levels of injury to the brain. An important factor in determining the level of neurological abnormality is the duration of the acceleration; longer periods of acceleration will result in traumatic coma whilst shorter periods will usually produce subdural hematoma. Loss of consciousness is a feature of all categories of traumatic brain injury (mild moderate or severe injury). In mild injury loss of consciousness persist for few minutes however in severe injury consciousness is lost from days to weeks. The duration of loss of consciousness is also directly related to reactive axons.
Contusions
Most TBI occur with contusions and a brain with contusions confirms the presence of brain injury as result of trauma. Contusions are also kinds of vascular injury particularly to small blood vessels. Different degrees of contusions exist following brain injury. Amongst them are those at right angles to the cortical surface. Contusions have several effects on the affected brain, typically it affects gyral crest and usually move towards a necrotic area including the sub adjacent white matter. Brain injury contusions have characteristics of continual expansion due to progressive hemorrhage, necrosis and edema. They are also referred to as coup contusions because they often occur beneath the impact site11-13. Other abnormalities that usually occur with brain contusions are skull fractures and lacerations where there is typical disruption of brain parenchyma. Any contusions that occur opposite to the impact site are termed contrecoup contusions.
Hemorrhage
Hemorrhage occurs in traumatic brain injury because of the tearing of blood vessels at the point of impact. However, there are also delayed post-traumatic hematomas that occur several hours after initial impact, which cause herniation and intracranial pressure elevation. Following brain injury, different kinds of bleeding may occur in different parts of the brain. The most common of vascular injury is subarachnoid hemorrhage, which is bleeding into the subarachnoid space. Subarachnoid hemorrhage is usually minor and may easily be absorbed into the cerebrospinal fluids. However, they can easily evolve into a space-occupying lesion.
Again, rupturing of bridging veins after an inertial acceleration of the brain leads to subdural hemorrhages that may extend over an entire hemisphere. Another frequent form of bleeding following head impact is intra-ventricular hemorrhage coupled with hematomas in the brain substance (intracerebral hematomas). All over the basal ganglia and central white matter can also be found hemorrhages called the intraparenchymal hematomas.
Lastly are the petechial hemorrhages that occur in diffuse vascular injury that leads to death within minutes after impact. Hematomas together with brain swelling contribute mainly to increase intracranial pressure, distortions, shifts and eventually herniation of brain parenchyma. Generally, edema occurs around brain contusions and intracerebral hematomas.
Axonal injury
Traumatic axonal injury can be categorized as primary axotomy or secondary axotomy. Primary axotomy occurs when there is complete resection of neural tissues along with glial cells and blood vessels whereas secondary axotomy occurs in less severe mechanical insults. In terms of duration, primary axotomy is an instantaneous process unlike secondary axotomy which takes many hours to days to occur14. Research has shown that between the period of primary axotomy and secondary axotomy lies a window of opportunity for therapeutic intervention15, 16. The help of immunocytochemistry can do detection of axonal injury in TBI where antibodies are deployed to transport proteins such as neurofilament protein, amyloid precursor protein (APP), and synaptophysin. APP staining is the most sensitive technique for axonal injury17 and it's able to detect axotomy within minutes after impact with an added advantage of minimum background interference since uninjured axons do not stain with APP18-22. Traumatic axonal injury displays several molecular changes to the brain. Early changes after axonal injury include influx of calcium either through receptor-mediated, voltage-mediated or transient defects in the plasmalemma. Calcium over load causes enzymes such as protease and gene activation manifested as axonal swelling. Complete axotomy occur and become apparent when wallerian degeneration occurs16, 23. Microglia reaction also occurs following axonal injury. Mechanical and ischemic injury contributes or combines to make up the total amount of axonal injury in a brain at any particular time. Staining techniques are however unable to distinguish between these two factors making up total axonal injury24.
Animal models and their types
To understand the basic elements of how the central nervous system responds to injury and mechanical inputs animal models have been the most reliable and significant techniques employed by researchers. The ultimate aims of most or all animal models is to reproduce and replicate as much as possible a clinical TBI in a laboratory setting depicting the morphological, biochemical, molecular and behavioral changes seen after TBI. There is a considerable amount of literature depicting several years of research that has been used to define the scenarios causing TBI in the laboratory using animal models. Research using animal models span on all scales from population-based surveys to the molecular level, all in an attempt to determine the significant signatures of injuries. To be able to maximize the efficacy and achieve useful outcomes and results from animal models for proper translation to the bedside certain clinical and salient points need to be considered and put into consideration. First of all the work must start from the clinical situation and environment to be able to define the incidence and prevalence of injury.
Finally and most importantly, all efforts and desire should be made to translate the laboratory outcomes to find effective countermeasures against insults caused by traumatic brain injury. Enormous amount of animal models of TBI have been developed since the early 1980s using different kinds of species including cats, dogs and nonhuman primates. Recent animal models of TBI have been developed using rodents and remain the most widely adopted species in preclinical studies. Rodents are dominating the TBI field of studies because of several reasons such as ethical issues elimination and easy postsurgical needs. Carry out research with rodents are also cost effective and simple compared to larger animals and human models. At present, several types of animal models of TBI exist, and the strengths and weakness of each model are discussed in table 2.
Animal models of TBI
| ANIMAL MODEL | STRENGTH | WEAKNESS |
|---|---|---|
| Repetitive Brain Injury Model | Effective in characterizing the molecular and cellular bases of repetitive injury. | Do not replicate the head movements, both the rotational and angular acceleration that are common in sports related injury116 |
| Blast Injury Model | Mimics the real morphological damage seen in the personnel who sustained TBI from the military conflicts results in the unique pathological features seen in blast-induced mild TBI | Do not replicate the posttraumatic seizures, a common consequence of blast - induced mild TBI seen in humans |
| Penetrating Ballistic-Like Brain Injury Model | helpful in characterizing the immediate and subacute (up to 7 days) changes in intracranial pressure seen after brain trauma 117.Captures several unique temporal aspects of a ballistic brain injury and may be a highly relevant model of moderate-to severe brain trauma for mechanistic studies. PBBI causes extensive intracerebral hemorrhage on the primary lesion site owing to the penetrating nature of the injury and the temporary cavity that it forms to truly model gunshot wound injuries to the brain. | Primary disadvantage concerns the expertise required of the investigator performing the procedure |
| Weight-Drop TBI Model | mimics closed head injury with accompanying concussion and contusion, a common type of TBI in humans. Inexpensive, easy to perform, and capable of producing graded diffuse axonal injury. | relatively high variability in injury severity that is produced |
| Controlled Cortical Impact Injury Model | time, velocity, and depth of impact can be controlled, making it more useful in studying the biomechanical changes that occur following TBI12. lack of rebound injury because the impact delivered by the device is gravity driven118. | only unilateral damage is produced, with rare involvement of the contralateral cortex |
| Fluid Percussion Injury Model | replicate the common pathophysiological features seen in human TBI119. Highly reproducible, and the investigator can regulate the severity of TBI. | does not produce skull fracture and results mainly in focal injury, it cannot replicate moderate-to severe TBI in humans where skull fractures and contusions across multiple brain regions are present120 . |
Physiological changes observed with animal model TBI
Animal models of TBI causes several physiological changes that are typical of clinical changes observed in human TBI. Categorically changes observed with animal models of TBI can be considered as either acute systemic, cerebrovascular, neurological or anatomical after injury. Acute systemic changes observed in some animals immediately after low levels of injury include an increased mean arterial blood pressure that remains at higher levels for the first 10-15minutes25 compared to the mean arterial blood pressure before injury. However, for a higher level of injury in some TBI models the laboratory species usually become hypotensive within an hour post injury. Nevertheless, in certain species tachycardia or bradycardia may be observed in the first minutes after injury.
Another prognostic factor that changes and is affected acutely in animal models is intracranial pressure. Intracranial pressure rises transiently within the first 20minutesto about 50mmHg25. Traumatic brain injury leads to an increase in central nervous system activity that will eventually compel plasma glucose levels to be elevated exponentially. Increased glucose levels tend to proportionally increase its utilization in the ventral tegmental nucleus of Gudden and the areas of the anterior thalamic nucleus26. These areas with increased metabolism are considered to be exhibiting functional changes of neural activity after injury.
A vital parameter that also increases transiently after traumatic brain injury is cerebral blood flow (CBF). The brain's inability to auto regulate CBF is related to endothelial lesions that appear after injury. Activation of processes that leads to Synthesis of prostaglandins and release of free radicals after mechanical injury to the brain pays a major role in these microvascular abnormalities. Studies have revealed also a transient break down in the blood brain barrier in vital anatomical parts of the brain especially in the brain stem regions after brain injury27, 28. Break in blood brain barrier allows for hemorrhages to occur as deep as pontomesencephalic junction25. At the clinical level measurements such as electroencephalography (EEG) are observed to vary after traumatic brain injury compared to the uninjured brain. EEG amplitude depresses acutely coupled with pupillary dilatations and apnea after brain injuries. The above physiological changes give insights into the respiratory, pulmonary, cardiovascular and cerebrovascular consequences of traumatic brain injury. Cerebrospinal fluid analysis following brain injury shows increased levels of acetylcholine which is a cholinergic neurotransmitter29. The clinical state of a patient depends on the levels of acetylcholine.
Significance of TBI animal models
By all standards our knowledge of the pathophysiology of TBI has been widened and broadened because of the development of clinically relevant experimental models of TBI30. Below we will elicit some of the pathophysiological findings that experimental TBI models have been able to reveal to scientist and researchers.
- Following TBI there be will be long term impairments of cognitive and neuromotor function31.
- There is a relationship between the extent of cognitive dysfunction and severity of TBI as well as neuronal loss32, 33.
- Neurological motor deficits shows signs of recovery within 1 year while cognitive deficits persists up to 1 year after severe TBI34.
- Axonal degeneration continues in the corpus callosum, striatum and injured cortex for as long as half a year and a year in the thalamus34.
- Ventriculomegaly, thalamic degeneration, shrinkage of the hippocampal pyramidal cell layer, progressive bilateral neuronal death in the dentate hilus, reactive astrocytosis, and progressive atrophy of the cortex, thalamus, hippocampus, and septum endures up to 1 year following brain injury35, 36.
- Substantial tissue loss occurs in the impact region after TBI.
- Neuronal cell loss happens in the hippocampus following TBI.
- A relationship tends to exist between neuronal cell loss and behavioral deficits12, 37-39.
- Cell death following TBI is primarily caused by necrosis40.
- Cell death occurs both in the acute and chronic phases after brain injury in neocortex, thalamus and hippocampus regions of the brain41.
- Primary and secondary axotomy both occur after traumatic brain injury however it is secondary axotomy that is widely regarded as the main pathological finding15, 42.
Limiting factors in TBI animal models
In the following sections, we will discuss some of the shortcomings or variables that investigators fail to incorporate into TBI animal models. These factors are extremely important in finding positive results from TBI animal models.
Secondary Insults
Secondary insults such as hypotension and hypoxemia occur following traumatic brain injury. Close to about 30% of severe TBI patients will exhibit hypotension and/or hypoxemia43. Even though the frequencies of secondary insults with TBI patients are comparatively high, animal models of TBI barely incorporate them. From the period of 1996 -2000 of the 168 cases of animal models of TBI reviewed in the journal of neurotrauma, only about 7 percent had incorporated a secondary insult7. Between secondary hypotension and secondary hypoxemia the later has been incorporated quiet often in animal models of TBI, technical ease being the most likely reason for this. A few rodent models of TBI have had posttraumatic hypotension been incorporated even so they do happen in conjunction with hypoxemia. Despite the fact that intracranial hypertension is a common factor associated with TBI in the clinic impact-acceleration models without secondary insults demonstrated no intracranial hypertension or neuronal death. Models that have tried to explain the effects of posttraumatic hypotension have done so more commonly in larger animals such as pigs and cats relatively to rodents. It is worth nothing that many and several studies have been conducted on the effects of fluid resuscitation strategies after traumatic brain injury44-50 while posttraumatic hypotensive effects have been poorly dealt with in the laboratory environment. Majority of these animal models of TBI have been able to include parameters such as intracranial pressure monitoring, cerebral blood flow, oxygen delivery and so on but very few have been able to be compared with secondary insults, hence it is easy to speculate that these models have been limited in their capacity to specifically explain the mechanisms of neuronal damage intensified by secondary insults leading to some of the failures pertaining to translating laboratory results to the bedside. This is not to say that fluid resuscitation models are not needed because optimal approach to fluid resuscitation for hypotensive patient following traumatic brain injury needs to be investigated. Studies have compared restoration of mean arterial pressure by either fluid restoration or treatment with the vasopressor; phenylephrine using a pig cerebral injury model and revealed that fluid resuscitation causes earlier cerebral blood flow51. In the other words restoration of mean arterial pressure alone will not counteract the many biochemical events of injury produced by hypotension.
Post traumatic Coma
Virtually all patients suffering from severe traumatic brain injury will go into coma. Posttraumatic coma is an important prognostic factor in quantifying mortality and morbidity52 after brain trauma. At the clinical level coma after the Glasgow Coma Scale (GCS) that ranges from a score of GCS3 - GCS15. This measures trauma important prognosticator is caused by several insults after brain injury. Compression of brain stem, diffuse axonal injury, and mass effect from hemorrhage are some of the etiologies of coma52. Incorporating coma into animal models is very crucial because GCS is able to predict the functional outcome of a traumatic brain injured patient. With an established fact that coma is such an important parameter for translating experimental results to the bedside modeling coma in laboratory animal models of TBI has proven to be tedious. The first successful model of post-traumatic coma in primates happened in 198253. The very few models that have shown some signs of hope have had their own form of limitations for example all the animals that were used to model posttraumatic coma by acceleration in the sagittal plane could not demonstrate DAI or coma but only concussive injury which is comma lasting for just a period of 15mins or less53. In an attempt to achieve better results with the modeling of posttraumatic coma in a laboratory setting acceleration in the coronal plane have been deployed; the end result to that approach was a coma lasting for more than 6 hours in half the population of animals used. Literature review shows that several attempts have been made to model this insult via modifications of the planes of acceleration that produces traumatic brain injury in a laboratory environment54. Although some knowledge and findings into posttraumatic coma have been actualized the duration and relationship between DAI and coma has been unsatisfactory55. DAI modeling has seen much success compared to coma. Several types of animal models (impact acceleration model, CCI, angular acceleration models) in larger animals have failed to produce coma56-58. The task involved in producing DAI with coma in TBI animal models may be connected to the difference in brain structure. Asphyxia insults have been used to make some advances in modeling coma using FPI however this does not accurately epitomize DAI -induced coma59, 60. In addition to the many means adopted to model traumatic coma in animal models mass effect has also been deployed61 and its proven futile in rodent models. Mechanical inputs alone is unable to produce coma in animal models of TBI, in the same vein unconsciousness has been produced without mechanical inputs62. The above varying result obtained from the experimental table has affected the advancement of therapeutic interventions in the clinic.
Neurointensive Care Monitoring and Management Strategies
Herniation of brain parenchyma as well as ischemia after TBI prompts clinicians to administer neurointensive care in an attempt to treat and improve patients' neurological deficits. Neurointensive care and monitoring is a function of both intracranial pressure (ICP) and cerebral perfusion pressure (CPP). The use of neurointensive strategies to counter the devastating consequences of ischemia has shown promising results63. Deeper understanding of ICP and CPP and the concept of neurointensive management are paramount for severe brain injury patients. Neurological deterioration after traumatic brain injury has been shown and demonstrated to be strongly linked to intracranial hypertension and ICP. The clinical significance of ICP is overwhelming but it is barely investigated in TBI models. Majority of models apply unclosed craniotomies that limit the monitoring of ICP. Also, those models with intact cranium often have skull fractures that also limit ICP monitoring. Although skull fractures and open craniotomies limit the monitoring of ICP and CPP they have been investigated and assessed at the acute phase in some laboratory TBI models64, 65,66. A couple of other means to monitor ICP/CPP indirectly has been conducted by investigating posttraumatic brain swelling/edema instead of ICP67, 68,69, 70 directly. Continual studies into the dynamics of ICP and CPP therapeutic interventions are required for the sub-acute period of traumatic brain injury. Compared to functional outcome investigations after traumatic brain injury very little has been done to discover the mechanisms underlying neurointensive care strategies after brain injury. Unfortunately, there are just few studies that target posttraumatic edema and intracranial hypertension. The overall severity of some animal models of TBI especially rodents is not entirely high so doesn't require mechanical ventilation and strict neurointensive care thus limiting the usefulness of these models in the clinical environment. The behavior of experimental models are therefore different in relation to the clinical settings as paradigms such as herniation and de-compressive craniotomies have not been effectively modeled61. Several other important aspects of neurointensive care that have poorly been addressed in TBI models include oxygen therapy, surgical decompression, catecholamine therapy and so on. Provision of oxygen after traumatic brain injury attenuates secondary ischemia and other secondary insults71 in the resuscitation phase. The absence of oxygen therapy in many experimental models of TBI has limited progress of novel therapeutic modalities in the clinical setting. Exogenous catecholamines that are administered to severe TBI patients to control mean arterial pressure (MAP) and CPP are also poorly investigated and understood. Models that have done some work in this regard have shown promising signs72, 73. Surgical de-compressive approaches that are common in the clinical setting have not been applied vigorously in TBI models. The size of infarct reduces significantly following de-compressive surgery74. Clinical trials have not been beneficial with several models lacking incorporation of surgical approach. It is possible that the lack of all these paradigms in experimental models has delineated clinically relevant strategies.
Recommended Clinical Secondary Insults Requiring Incorporation into TBI Animal Models
To achieve better results from laboratory TBI animal models we will recommend insertion of some of the variables depicted in table 3 in future research.
Classification of secondary brain insults.
| Systemic Insults | Ischaemia | hypotension |
| anaemia or changes in haemodynamics | ||
| Hypoxemia/ Hypercarbia | respiratory obstruction | |
| Pulmonary complications | ||
| suppression of respiratory | ||
| Hyperthermia | post-traumatic cerebral inflammation | |
| thrombophlebitis | ||
| drug reaction | ||
| direct hypothalamic damage | ||
| Electrolyte abnormalities | hypernatremia | |
| hypernatremia | ||
| hypomagnesaemia | ||
| hypocalcaemia | ||
| Hyperglycaemia | anaerobic metabolism | |
| increase inflammation response | ||
| aggravate brain ischaemia | ||
| Polytrauma | bone fracture | |
| liver laceration | ||
| other organs injury | ||
| Infection/Sepsis | increase brain inflammation | |
| brain metabolic alterations | ||
| Thrombocytopenia/ Coagulopathy | new or progressive haemorrhagic changes | |
| Ethanol consumption | abnormal haemodynamic response | |
| Excessive release of BNP | ||
| suppression of ADH | ||
| Intracranial Insults | Increased ICP/ Cerebral hypoperfusion | |
| Brain shift/Herniation | ||
| Brain oedema/swelling | ||
| Cerebral angiospasm | ||
| Hydrocephalus | ||
| Epilepsy |
Hyperglycemia
Hyperglycemia is one of the most reliable prognosticator after traumatic brain injury75, 76. Hyperglycemia causes lactic acidosis and neuronal injury in the brain by influencing anaerobic metabolism and production of excitatory amino acids. Even though there isn't a specific standard glucose level; clinicians adopt strict glucose control measures after traumatic brain injury to reduce devastating effects of hyperglycemia77. For such an important clinical factor without a specific or standardized guideline it is paramount that clinicians and researchers delve deeper to acquire more knowledge. In creating TBI models that will cater for hyperglycemia and its mechanistic effect one should consider TBI severity as well as the amount of glucose infusion. Currently TBI models complicated by hyperglycemia have had inconsistent results with some studies stating that hyperglycemia has a negative outcome on TBI patients whilst others have stated otherwise78-81. We suggest that the timing of glucose infusion after or before TBI should be further investigated. Secondly different combinations of time of glucose infusion and TBI severity needs to be assessed to gain much understanding about hyperglycemia and TBI correlations. Hyperglycemia does not cause adverse effects after traumatic brain injury without other secondary insults such as ischemia82 hence TBI models complicated by hyperglycemia will be appropriate to be further complicated with ischemia for effectiveness and clinical importance.
Hyponatremia
TBI is often associated with electrolyte abnormalities including hypernatremia and hyponatremia. Hyponatremia tends to have a much higher prognostic effect on TBI outcomes compared to hypernatremia83. Hyponatremia can be classified as either mild or severe with different clinical outcomes. It's been proposed that hypopituitarism, volume overload, syndrome of inappropriate secretion of anti-diuretic hormone (SIADH) and cerebral salt wasting syndrome (CSWS) cause posttraumatic hyponatremia84. TBI models complicated with hyponatremia require further research so as to be able to answer such critical questions:
1) What is the real mechanism underlying the causes of hyponatremia after TBI?
2) Is hyponatremia just a symptom or it's an insult leading to poor outcomes after TBI?
Even though thyponatremia is a common insult observed in TBI patients they have not been really imposed on laboratory TBI models. Previous work on hyponatremia and TBI could not clarify the pathogenesis of how hyponatremia aggravates brain injury85. Another study has linked the adverse effects of hyponatremia to hypoxic or ischemic factors instead of distorted blood brain barrier86. Another avenue that could be deployed to model hyponatremia in TBI model is relying on the idea that hyponatremia results from hypopituitarism87.
Last but not the least infusion of ethanol to TBI models can also be sorted to reproduce hyponatremia88. Should some of these avenues be investigated further and deeper there is greater chance that some of the laboratory results could be translated to the bed side for effective therapies against TBI.
Hyperthermia
Post-traumatic hyperthermia is a state of body temperatures without background infections that is higher than normothermic patients. Basically, temperature elevations after trauma are cause by inflammations, or direct hypothalamic provocations. Posttraumatic hyperthermia is a neurogenic fever that is comparatively resistant to antipyretic drugs89, 90. Irrespective of the causes of hyperthermia, it periodically causes high metabolism as well as high levels of leukocyte activation. The cumulative effects of posttraumatic hyperthermia worsen the outcome of patients90, 91. Traumatic brain injury patients require to be maintained at a normal core temperature92. This review aims to discuss post-traumatic hyperthermia since its pathophysiology is poorly understood both at the experimental and clinical levels. Moreover current strategies for controlling post-traumatic hyperthermia remain ineffective and unreliable92. Some research conducted in the past about TBI linked the damaging of hypothalamus to hyperthermia90, 93. Efforts to incorporate hyperthermia into TBI models so as to help understand the mechanisms underlying why hyperthermia aggravates neuronal damage and loss has also been conducted94 and found promising results. Post-traumatic hyperthermia was however incorporated at different periods after CCI or FPI (delayed94 and immediately95, 96). Special attention should be paid to investigating both core and brain temperature after traumatic brain injury since both plays critical role in neutrophils elevation. Whole body hyperthermia leads to the elevation of neurotoxic zinc affecting the hippocampal neurons97, 98 whiles cortical hyperthermia increases extracellular glutamate levels99. These and many other results buttress the suggestion that posttraumatic hyperthermia increases and induces inflammation after TBI.
To further the advancement of clinical therapies involving TBI patients, researchers will have to focus their attention on incorporating posttraumatic hyperthermia in their animal models for all kinds of TBI grading / severities (mild, moderate and severe).
Hypoxemia/hypotension/ischemia
The brain by default is an organ with high rate of metabolism which increases further after traumatic brain injury100. An increase metabolism requires high oxygen supply to meet the demands however the brain lacks oxygen reservoir hence it is extremely sensitive to hypoxemia. In addition to high rates of metabolism other insults that causes hypoxemia following traumatic brain injury includes pulmonary disorders such as pneumothorax or atelectasis101.
Finally, respiratory failure as a result of disrupted brain pathways also contributes to brain hypoxemia after TBI. Mortality and morbidity rates are significantly affected by hypoxemia or hypotension after traumatic brain injury91, 102. The significance of hypoxemia as far as finding therapeutic interventions of clinical TBI cannot be overemphasized for the very reason that severe hypoxemia increases the neurological deficits observed after traumatic brain injury as well as the aggravation of the injured brain103. Correlations of hypoxemia with high energy phosphate104, cerebral blood flow105 and blood brain barrier dysfunction105 proves that TBI models require the incorporation of hypoxemia to increase their efficacy and clinical significance. Other factors that require attention when incorporating hypoxemia into TBI models include the connection between hypoxia and the severity of TBI. In building and developing TBI models it is recommended that arterial oxygen pressure be maintained at a steady rate and efforts made to set the FiO2 at 21%. Should these factors and issues be dealt with appropriately when developing animal TBI models several adverse effects as a result of hypoxemia can be displayed and understood101, 106-111. Experimental studies show that hypoxemia has more adverse effects than hypotension after TBI106, 112-114 whilst clinical studies have suggested hypotension to be the sole prognosticator after TBI91. These conflicting results warrant further research and investigation into the synergistic effect of hypoxemia and hypotension43, 114, 115.
Physiology of traumatic brain injury
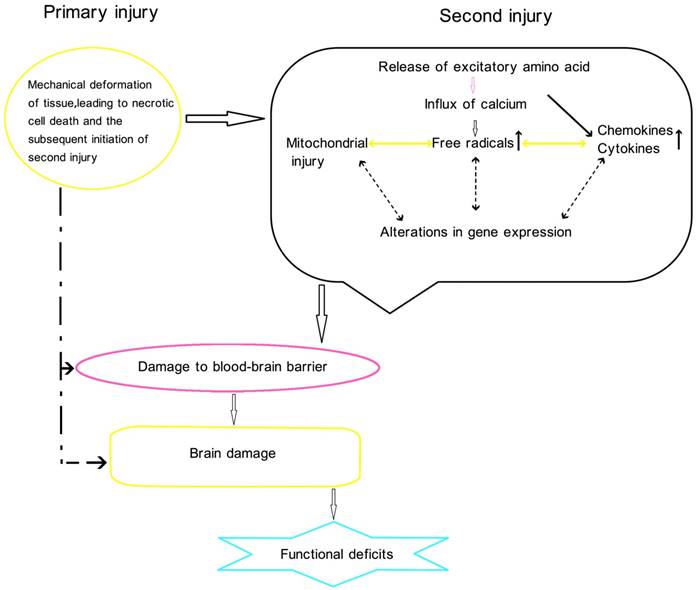
Conclusion
Several TBI models has assisted in explaining the molecular and biochemical basis of traumatic brain injuries to a greater extend this has helped in finding treatments to intervene the cascades of events that occur after primary injury to the brain. Despite all the carefully conducted TBI researches; damaging insults to the brain as a result of trauma still continues to be a greater burden to clinician-scientist. This review has sighted that the discrepancies in translating bench results to the bedside could be due to the lack of secondary insults in laboratory TBI models. Future models of TBI should make necessary effort to incorporate secondary insults to achieve as close as possible clinical scenarios in the laboratory.
Abbreviations
TBI: traumatic brain injury; ICP: intracranial pressure; CT: computer topography; MRI: magnetic resonance imaging; DAI: Diffuse axonal injury; DVI: Diffuse vascular injury; APP: amyloid precursor protein; PBBI: Penetrating Ballistic-Like Brain Injury; CBF: cerebral blood flow; EEG: electroencephalography; GCS: Glasgow Coma Scale; CCI: cortical compact injury; CPP: cerebral perfusion pressure; SIADH: syndrome of inappropriate secretion of anti-diuretic hormone.
Acknowledgements
This work was supported by the National Science Funding of China (81641045 and 81371396), Natural Science Funding of Zhejiang Province (LQ15H090006), Zhejiang Key Health Science and Technology Project (WKJ2013-2-022), Zhejiang Health Science and Technology Project (2016RCA022), Wenzhou City Science and Technology Project (Y20150042).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Reilly P, Bullock R. Head injury: Pathophysiology and management of severe closed injury. London; New York: Chapman & Hall Medical. 1997
2. Graham DI, Ford I, Adams JH, Doyle D, Lawrence AE, McLellan DR. et al. Fatal head injury in children. Journal of clinical pathology. 1989;42:18-22
3. Blumbergs PC, Jones NR, North JB. Diffuse axonal injury in head trauma. Journal of neurology, neurosurgery, and psychiatry. 1989;52:838-841
4. Bullock MR, Lyeth BG, Muizelaar JP. Current status of neuroprotection trials for traumatic brain injury: Lessons from animal models and clinical studies. Neurosurgery. 1999;45:207-217 discussion 217-220
5. Cohadon F. Brain protection. Advances and technical standards in neurosurgery. 1994;21:77-152
6. Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and nmda receptors in traumatic brain injury. Science. 1989;244:798-800
7. Statler KD, Jenkins LW, Dixon CE, Clark RS, Marion DW, Kochanek PM. The simple model versus the super model: Translating experimental traumatic brain injury research to the bedside. Journal of neurotrauma. 2001;18:1195-1206
8. Gennarelli TA. Mechanisms of brain injury. The Journal of emergency medicine. 1993;11(Suppl 1):5-11
9. Armonda RA, Bell RS, Vo AH, Ling G, DeGraba TJ, Crandall B. et al. Wartime traumatic cerebral vasospasm: Recent review of combat casualties. Neurosurgery. 2006;59:1215-1225 discussion 1225
10. Ling G, Bandak F, Armonda R, Grant G, Ecklund J. Explosive blast neurotrauma. Journal of neurotrauma. 2009;26:815-825
11. Cortez SC, McIntosh TK, Noble LJ. Experimental fluid percussion brain injury: Vascular disruption and neuronal and glial alterations. Brain research. 1989;482:271-282
12. Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. Journal of neuroscience methods. 1991;39:253-262
13. McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H. et al. Traumatic brain injury in the rat: Characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233-244
14. Maxwell WL, Kosanlavit R, McCreath BJ, Reid O, Graham DI. Freeze-fracture and cytochemical evidence for structural and functional alteration in the axolemma and myelin sheath of adult guinea pig optic nerve fibers after stretch injury. Journal of neurotrauma. 1999;16:273-284
15. Povlishock JT, Christman CW. The pathobiology of traumatically induced axonal injury in animals and humans: A review of current thoughts. Journal of neurotrauma. 1995;12:555-564
16. Povlishock JT, Hayes RL, Michel ME, McIntosh TK. Workshop on animal models of traumatic brain injury. Journal of neurotrauma. 1994;11:723-732
17. Sherriff FE, Bridges LR, Gentleman SM, Sivaloganathan S, Wilson S. Markers of axonal injury in post mortem human brain. Acta neuropathologica. 1994;88:433-439
18. Finnie JW, Blumbergs PC, Manavis J, Summersides GE, Davies RA. Evaluation of brain damage resulting from penetrating and non-penetrating captive bolt stunning using lambs. Australian veterinary journal. 2000;78:775-778
19. Finnie JW, Van den Heuvel C, Gebski V, Manavis J, Summersides GE, Blumbergs PC. Effect of impact on different regions of the head of lambs. Journal of comparative pathology. 2001;124:159-164
20. Lewis SB, Finnie JW, Blumbergs PC, Scott G, Manavis J, Brown C. et al. A head impact model of early axonal injury in the sheep. Journal of neurotrauma. 1996;13:505-514
21. Van den Heuvel C, Blumbergs PC, Finnie JW, Manavis J, Jones NR, Reilly PL. et al. Upregulation of amyloid precursor protein messenger rna in response to traumatic brain injury: An ovine head impact model. Experimental neurology. 1999;159:441-450
22. Van Den Heuvel C, Blumbergs P, Finnie J, Manavis J, Lewis S, Jones N. et al. Upregulation of amyloid precursor protein and its mrna in an experimental model of paediatric head injury. Journal of clinical neuroscience: official journal of the Neurosurgical Society of Australasia. 2000;7:140-145
23. Povlishock JT, Marmarou A, McIntosh T, Trojanowski JQ, Moroi J. Impact acceleration injury in the rat: Evidence for focal axolemmal change and related neurofilament sidearm alteration. Journal of neuropathology and experimental neurology. 1997;56:347-359
24. Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ. Staining of amyloid precursor protein to study axonal damage in mild head injury. Lancet. 1994;344:1055-1056
25. Sullivan HG, Martinez J, Becker DP, Miller JD, Griffith R, Wist AO. Fluid-percussion model of mechanical brain injury in the cat. Journal of neurosurgery. 1976;45:521-534
26. Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD. et al. The [14c]deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat. Journal of neurochemistry. 1977;28:897-916
27. Povlishock JT, Becker DP, Miller JD, Jenkins LW, Dietrich WD. The morphopathologic substrates of concussion? Acta neuropathologica. 1979;47:1-11
28. Povlishock JT, Becker DP, Sullivan HG, Miller JD. Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain research. 1978;153:223-239
29. Bornstein MB. Presence and action of acetylcholine in experimental brain trauma. Journal of neurophysiology. 1946;9:349-366
30. Laurer HL, McIntosh TK. Experimental models of brain trauma. Current opinion in neurology. 1999;12:715-721
31. Levin HS, Gary HE Jr, Eisenberg HM, Ruff RM, Barth JT, Kreutzer J. et al. Neurobehavioral outcome 1 year after severe head injury. Experience of the traumatic coma data bank. Journal of neurosurgery. 1990;73:699-709
32. Smith DH, Okiyama K, Thomas MJ, Claussen B, McIntosh TK. Evaluation of memory dysfunction following experimental brain injury using the morris water maze. Journal of neurotrauma. 1991;8:259-269
33. Hicks RR, Smith DH, Lowenstein DH, Saint Marie R, McIntosh TK. Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. Journal of neurotrauma. 1993;10:405-414
34. Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359-369
35. Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta neuropathologica. 2002;103:607-614
36. Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI. et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. Journal of neurotrauma. 1997;14:715-727
37. Fox GB, Fan L, Levasseur RA, Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. Journal of neurotrauma. 1998;15:599-614
38. Hamm RJ, Dixon CE, Gbadebo DM, Singha AK, Jenkins LW, Lyeth BG. et al. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. Journal of neurotrauma. 1992;9:11-20
39. Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF. et al. A model of parasagittal controlled cortical impact in the mouse: Cognitive and histopathologic effects. Journal of neurotrauma. 1995;12:169-178
40. Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: A light and electron microscopic study in rats. Journal of neurotrauma. 1994;11:289-301
41. Schwab JM, Brechtel K, Conrad S, Schluesener HJ. From cell death to neuronal regeneration: Building a new brain after traumatic brain injury (j neuropathol exp neurol 2003;62:801-11. Journal of neuropathology and experimental neurology. 2004;63:180-181
42. Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: A review. Journal of neurotrauma. 1997;14:419-440
43. Chesnut RM, Marshall LF, Klauber MR, Blunt BA, Baldwin N, Eisenberg HM. et al. The role of secondary brain injury in determining outcome from severe head injury. The Journal of trauma. 1993;34:216-222
44. Bourguignon PR, Shackford SR, Shiffer C, Nichols P, Nees AV. Delayed fluid resuscitation of head injury and uncontrolled hemorrhagic shock. Archives of surgery. 1998;133:390-398
45. DeWitt DS, Prough DS, Deal DD, Vines SM, Hoen H. Hypertonic saline does not improve cerebral oxygen delivery after head injury and mild hemorrhage in cats. Critical care medicine. 1996;24:109-117
46. Glass TF, Fabian MJ, Schweitzer JB, Weinberg JA, Proctor KG. Secondary neurologic injury resulting from nonhypotensive hemorrhage combined with mild traumatic brain injury. Journal of neurotrauma. 1999;16:771-782
47. Ramming S, Shackford SR, Zhuang J, Schmoker JD. The relationship of fluid balance and sodium administration to cerebral edema formation and intracranial pressure in a porcine model of brain injury. The Journal of trauma. 1994;37:705-713
48. Shackford SR. Effect of small-volume resuscitation on intracranial pressure and related cerebral variables. The Journal of trauma. 1997;42:S48-53
49. Stern SA, Zink BJ, Mertz M, Wang X, Dronen SC. Effect of initially limited resuscitation in a combined model of fluid-percussion brain injury and severe uncontrolled hemorrhagic shock. Journal of neurosurgery. 2000;93:305-314
50. Zhuang J, Shackford SR, Schmoker JD, Pietropaoli JA Jr. Colloid infusion after brain injury: Effect on intracranial pressure, cerebral blood flow, and oxygen delivery. Critical care medicine. 1995;23:140-148
51. Alspaugh DM, Sartorelli K, Shackford SR, Okum EJ, Buckingham S, Osler T. Prehospital resuscitation with phenylephrine in uncontrolled hemorrhagic shock and brain injury. The Journal of trauma. 2000;48:851-863 discussion 863-854
52. Gennarelli TA. Head injury in man and experimental animals: Clinical aspects. Acta neurochirurgica. Supplementum. 1983;32:1-13
53. Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, Marcincin RP. Diffuse axonal injury and traumatic coma in the primate. Annals of neurology. 1982;12:564-574
54. Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D. et al. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. Journal of neurosurgery. 2000;93:315-322
55. Smith DH, Chen XH, Xu BN, McIntosh TK, Gennarelli TA, Meaney DF. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. Journal of neuropathology and experimental neurology. 1997;56:822-834
56. Foda MA, Marmarou A. A new model of diffuse brain injury in rats. Part ii: Morphological characterization. Journal of neurosurgery. 1994;80:301-313
57. Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats. Part i: Pathophysiology and biomechanics. Journal of neurosurgery. 1994;80:291-300
58. Xiao-Sheng H, Sheng-Yu Y, Xiang Z, Zhou F, Jian-ning Z. Diffuse axonal injury due to lateral head rotation in a rat model. Journal of neurosurgery. 2000;93:626-633
59. Katz L, Ebmeyer U, Safar P, Radovsky A, Neumar R. Outcome model of asphyxial cardiac arrest in rats. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1995;15:1032-1039
60. Glass TF, Fabian MJ, Schweitzer JB, Weinberg JA, Proctor KG. The impact of hypercarbia on the evolution of brain injury in a porcine model of traumatic brain injury and systemic hemorrhage. Journal of neurotrauma. 2001;18:57-71
61. Pomeranz S, Safar P, Radovsky A, Tisherman SA, Alexander H, Stezoski W. The effect of resuscitative moderate hypothermia following epidural brain compression on cerebral damage in a canine outcome model. Journal of neurosurgery. 1993;79:241-251
62. Hayes RL, Pechura CM, Katayama Y, Povlishock JT, Giebel ML, Becker DP. Activation of pontine cholinergic sites implicated in unconsciousness following cerebral concussion in the cat. Science. 1984;223:301-303
63. Rosner MJ, Rosner SD, Johnson AH. Cerebral perfusion pressure: Management protocol and clinical results. Journal of neurosurgery. 1995;83:949-962
64. Cherian L, Robertson CS, Contant CF Jr, Bryan RM Jr. Lateral cortical impact injury in rats: Cerebrovascular effects of varying depth of cortical deformation and impact velocity. Journal of neurotrauma. 1994;11:573-585
65. Kroppenstedt SN, Kern M, Thomale UW, Schneider GH, Lanksch WR, Unterberg AW. Effect of cerebral perfusion pressure on contusion volume following impact injury. Journal of neurosurgery. 1999;90:520-526
66. Mirski AM, Denchev ID, Schnitzer SM, Hanley FD. Comparison between hypertonic saline and mannitol in the reduction of elevated intracranial pressure in a rodent model of acute cerebral injury. Journal of neurosurgical anesthesiology. 2000;12:334-344
67. Bareyre F, Wahl F, McIntosh TK, Stutzmann JM. Time course of cerebral edema after traumatic brain injury in rats: Effects of riluzole and mannitol. Journal of neurotrauma. 1997;14:839-849
68. Feldman Z, Zachari S, Reichenthal E, Artru AA, Shapira Y. Brain edema and neurological status with rapid infusion of lactated ringer's or 5% dextrose solution following head trauma. Journal of neurosurgery. 1995;83:1060-1066
69. Talmor D, Shapira Y, Artru AA, Gurevich B, Merkind V, Katchko L. et al. 0.45% saline and 5% dextrose in water, but not 0.9% saline or 5% dextrose in 0.9% saline, worsen brain edema two hours after closed head trauma in rats. Anesthesia and analgesia. 1998;86:1225-1229
70. Whalen MJ, Carlos TM, Dixon CE, Robichaud P, Clark RS, Marion DW. et al. Reduced brain edema after traumatic brain injury in mice deficient in p-selectin and intercellular adhesion molecule-1. Journal of leukocyte biology. 2000;67:160-168
71. Menzel M, Doppenberg EM, Zauner A, Soukup J, Reinert MM, Bullock R. Increased inspired oxygen concentration as a factor in improved brain tissue oxygenation and tissue lactate levels after severe human head injury. Journal of neurosurgery. 1999;91:1-10
72. McIntosh TK, Yu T, Gennarelli TA. Alterations in regional brain catecholamine concentrations after experimental brain injury in the rat. Journal of neurochemistry. 1994;63:1426-1433
73. Feeney DM, Weisend MP, Kline AE. Noradrenergic pharmacotherapy, intracerebral infusion and adrenal transplantation promote functional recovery after cortical damage. Journal of neural transplantation & plasticity. 1993;4:199-213
74. Engelhorn T, Doerfler A, Kastrup A, Beaulieu C, de Crespigny A, Forsting M. et al. Decompressive craniectomy, reperfusion, or a combination for early treatment of acute "malignant" cerebral hemispheric stroke in rats? Potential mechanisms studied by mri. Stroke; a journal of cerebral circulation. 1999;30:1456-1463
75. Muehlschlegel S, Carandang R, Ouillette C, Hall W, Anderson F, Goldberg R. Frequency and impact of intensive care unit complications on moderate-severe traumatic brain injury: Early results of the outcome prognostication in traumatic brain injury (optimism) study. Neurocritical care. 2013;18:318-331
76. Rovlias A, Kotsou S. The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery. 2000;46:335-342 discussion 342-333
77. Liu-DeRyke X, Collingridge DS, Orme J, Roller D, Zurasky J, Rhoney DH. Clinical impact of early hyperglycemia during acute phase of traumatic brain injury. Neurocritical care. 2009;11:151-157
78. Cherian L, Hannay HJ, Vagner G, Goodman JC, Contant CF, Robertson CS. Hyperglycemia increases neurological damage and behavioral deficits from post-traumatic secondary ischemic insults. Journal of neurotrauma. 1998;15:307-321
79. Kraft SA, Larson CP Jr, Shuer LM, Steinberg GK, Benson GV, Pearl RG. Effect of hyperglycemia on neuronal changes in a rabbit model of focal cerebral ischemia. Stroke; a journal of cerebral circulation. 1990;21:447-450
80. Wang YY, Chen CJ, Lin SY, Chuang YH, Sheu WH, Tung KC. Hyperglycemia is associated with enhanced gluconeogenesis in a rat model of permanent cerebral ischemia. Molecular and cellular endocrinology. 2013;367:50-56
81. Vink R, Golding EM, Williams JP, McIntosh TK. Blood glucose concentration does not affect outcome in brain trauma: A 31p mrs study. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1997;17:50-53
82. Cherian L, Goodman JC, Robertson CS. Effect of glucose administration on contusion volume after moderate cortical impact injury in rats. Journal of neurotrauma. 1998;15:1059-1066
83. Van Beek JG, Mushkudiani NA, Steyerberg EW, Butcher I, McHugh GS, Lu J. et al. Prognostic value of admission laboratory parameters in traumatic brain injury: Results from the impact study. Journal of neurotrauma. 2007;24:315-328
84. Moro N, Katayama Y, Igarashi T, Mori T, Kawamata T, Kojima J. Hyponatremia in patients with traumatic brain injury: Incidence, mechanism, and response to sodium supplementation or retention therapy with hydrocortisone. Surgical neurology. 2007;68:387-393
85. Ke C, Poon WS, Ng HK, Tang NL, Chan Y, Wang JY. et al. The impact of acute hyponatraemia on severe traumatic brain injury in rats. Acta neurochirurgica. Supplement. 2000;76:405-408
86. Adler S, Williams D, Verbalis JG. Effect of acute and chronic hyponatremia on blood-brain barrier function in the rat. NMR in biomedicine. 1993;6:119-124
87. Dohanics J, Hoffman GE, Verbalis JG. Chronic hyponatremia reduces survival of magnocellular vasopressin and oxytocin neurons after axonal injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1996;16:2373-2380
88. Katada R, Nishitani Y, Honmou O, Okazaki S, Houkin K, Matsumoto H. Prior ethanol injection promotes brain edema after traumatic brain injury. Journal of neurotrauma. 2009;26:2015-2025
89. Agrawal A, Timothy J, Thapa A. Neurogenic fever. Singapore medical journal. 2007;48:492-494
90. Thompson HJ, Tkacs NC, Saatman KE, Raghupathi R, McIntosh TK. Hyperthermia following traumatic brain injury: A critical evaluation. Neurobiology of disease. 2003;12:163-173
91. McHugh GS, Engel DC, Butcher I, Steyerberg EW, Lu J, Mushkudiani N. et al. Prognostic value of secondary insults in traumatic brain injury: Results from the impact study. Journal of neurotrauma. 2007;24:287-293
92. Cairns CJ, Andrews PJ. Management of hyperthermia in traumatic brain injury. Current opinion in critical care. 2002;8:106-110
93. Rudy TA, Williams JW, Yaksh TL. Antagonism by indomethacin of neurogenic hyperthermia produced by unilateral puncture of the anterior hypothalamic/preoptic region. The Journal of physiology. 1977;272:721-736
94. Dietrich WD, Alonso O, Halley M, Busto R. Delayed posttraumatic brain hyperthermia worsens outcome after fluid percussion brain injury: A light and electron microscopic study in rats. Neurosurgery. 1996;38:533-541 discussion 541
95. Whalen MJ, Carlos TM, Clark RS, Marion DW, DeKosky MS, Heineman S. et al. The relationship between brain temperature and neutrophil accumulation after traumatic brain injury in rats. Acta neurochirurgica. Supplement. 1997;70:260-261
96. Chatzipanteli K, Alonso OF, Kraydieh S, Dietrich WD. Importance of posttraumatic hypothermia and hyperthermia on the inflammatory response after fluid percussion brain injury: Biochemical and immunocytochemical studies. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2000;20:531-542
97. Suh SW, Frederickson CJ, Danscher G. Neurotoxic zinc translocation into hippocampal neurons is inhibited by hypothermia and is aggravated by hyperthermia after traumatic brain injury in rats. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2006;26:161-169
98. Tsuchiya D, Hong S, Suh SW, Kayama T, Panter SS, Weinstein PR. Mild hypothermia reduces zinc translocation, neuronal cell death, and mortality after transient global ischemia in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2002;22:1231-1238
99. Adachi H, Fujisawa H, Maekawa T, Yamashita T, Ito H. Changes in the extracellular glutamate concentrations in the rat cortex following localized by hyperthermia. International journal of hyperthermia: the official journal of European Society for Hyperthermic Oncology, North American Hyperthermia Group. 1995;11:587-599
100. Cormio M, Robertson CS, Narayan RK. Secondary insults to the injured brain. Journal of clinical neuroscience: official journal of the Neurosurgical Society of Australasia. 1997;4:132-148
101. Hellewell SC, Yan EB, Agyapomaa DA, Bye N, Morganti-Kossmann MC. Post-traumatic hypoxia exacerbates brain tissue damage: Analysis of axonal injury and glial responses. Journal of neurotrauma. 2010;27:1997-2010
102. Stahel PF, Smith WR, Moore EE. Hypoxia and hypotension, the "lethal duo" in traumatic brain injury: Implications for prehospital care. Intensive care medicine. 2008;34:402-404
103. Ishige N, Pitts LH, Hashimoto T, Nishimura MC, Bartkowski HM. Effect of hypoxia on traumatic brain injury in rats: Part 1. Changes in neurological function, electroencephalograms, and histopathology. Neurosurgery. 1987;20:848-853
104. Ishige N, Pitts LH, Pogliani L, Hashimoto T, Nishimura MC, Bartkowski HM. et al. Effect of hypoxia on traumatic brain injury in rats: Part 2. Changes in high energy phosphate metabolism. Neurosurgery. 1987;20:854-858
105. Ishige N, Pitts LH, Berry I, Carlson SG, Nishimura MC, Moseley ME. et al. The effect of hypoxia on traumatic head injury in rats: Alterations in neurologic function, brain edema, and cerebral blood flow. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 1987;7:759-767
106. Clark RS, Kochanek PM, Dixon CE, Chen M, Marion DW, Heineman S. et al. Early neuropathologic effects of mild or moderate hypoxemia after controlled cortical impact injury in rats. Journal of neurotrauma. 1997;14:179-189
107. Chen M, Clark RS, Kochanek PM, Chen J, Schiding JK, Stetler RA. et al. 72-kda heat shock protein and mrna expression after controlled cortical impact injury with hypoxemia in rats. Journal of neurotrauma. 1998;15:171-181
108. Feng JF, Zhao X, Gurkoff GG, Van KC, Shahlaie K, Lyeth BG. Post-traumatic hypoxia exacerbates neuronal cell death in the hippocampus. Journal of neurotrauma. 2012;29:1167-1179
109. Bauman RA, Widholm JJ, Petras JM, McBride K, Long JB. Secondary hypoxemia exacerbates the reduction of visual discrimination accuracy and neuronal cell density in the dorsal lateral geniculate nucleus resulting from fluid percussion injury. Journal of neurotrauma. 2000;17:679-693
110. Dave JR, Bauman RA, Long JB. Hypoxia potentiates traumatic brain injury-induced expression of c-fos in rats. Neuroreport. 1997;8:395-398
111. Yan EB, Hellewell SC, Bellander BM, Agyapomaa DA, Morganti-Kossmann MC. Post-traumatic hypoxia exacerbates neurological deficit, neuroinflammation and cerebral metabolism in rats with diffuse traumatic brain injury. Journal of neuroinflammation. 2011;8:147
112. Clark RS, Chen J, Watkins SC, Kochanek PM, Chen M, Stetler RA. et al. Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:9172-9182
113. Navarro JC, Pillai S, Cherian L, Garcia R, Grill RJ, Robertson CS. Histopathological and behavioral effects of immediate and delayed hemorrhagic shock after mild traumatic brain injury in rats. Journal of neurotrauma. 2012;29:322-334
114. Dennis AM, Haselkorn ML, Vagni VA, Garman RH, Janesko-Feldman K, Bayir H. et al. Hemorrhagic shock after experimental traumatic brain injury in mice: Effect on neuronal death. Journal of neurotrauma. 2009;26:889-899
115. Foley LM, Iqbal O'Meara AM, Wisniewski SR, Hitchens TK, Melick JA, Ho C. et al. Mri assessment of cerebral blood flow after experimental traumatic brain injury combined with hemorrhagic shock in mice. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:129-136
116. Guskiewicz KM, Mihalik JP. Biomechanics of sport concussion: Quest for the elusive injury threshold. Exercise and sport sciences reviews. 2011;39:4-11
117. Wei G, Lu XC, Yang X, Tortella FC. Intracranial pressure following penetrating ballistic-like brain injury in rats. Journal of neurotrauma. 2010;27:1635-1641
118. Lighthall JW. Controlled cortical impact: A new experimental brain injury model. Journal of neurotrauma. 1988;5:1-15
119. Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA. et al. Lateral fluid percussion brain injury: A 15-year review and evaluation. Journal of neurotrauma. 2005;22:42-75
120. Hardman JM, Manoukian A. Pathology of head trauma. Neuroimaging clinics of North America. 2002;12:175-187 vii
Author contact
![]() Corresponding authors: Qichuan ZhuGe, M.D., Department of Neurosurgery, First Affiliated Hospital, Wenzhou Medical University, Wenzhou, China Tel: 86-577-55578085 Fax: 86-577-55578033 Email: zhugeqichuan163.com; Lijie Huang, M.D., Ph.D., Department of Neurosurgery, First Affiliated Hospital, Wenzhou Medical University, Wenzhou, China Tel: 86-577-55579352 Fax: 86-577-55578999 Email: lijiehuangwycom
Corresponding authors: Qichuan ZhuGe, M.D., Department of Neurosurgery, First Affiliated Hospital, Wenzhou Medical University, Wenzhou, China Tel: 86-577-55578085 Fax: 86-577-55578033 Email: zhugeqichuan163.com; Lijie Huang, M.D., Ph.D., Department of Neurosurgery, First Affiliated Hospital, Wenzhou Medical University, Wenzhou, China Tel: 86-577-55579352 Fax: 86-577-55578999 Email: lijiehuangwycom