Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(5):484-493. doi:10.7150/ijms.18059 This issue Cite
Research Paper
Effects of C-reactive protein on the expression of matrix metalloproteinases and their inhibitors via Fcγ receptors on 3T3-L1 adipocytes
Kumiko Nakai1, 2, Hideki Tanaka1, 2, Kazuhiro Yamanaka1, Yumi Takahashi3, Fumiko Murakami3, Rieko Matsuike3, Jumpei Sekino3, Natsuko Tanabe2, 4, Toyoko Morita1, 5, Yoji Yamazaki5, Takayuki Kawato1, 2 ![]() , Masao Maeno1, 2
, Masao Maeno1, 2
1. Department of Oral Health Sciences, Nihon University School of Dentistry, Tokyo, Japan;
2. Division of Functional Morphology, Dental Research Center, Nihon University School of Dentistry, Tokyo, Japan;
3. Nihon University Graduate School of Dentistry, Tokyo, Japan;
4. Department of Biochemistry, Nihon University School of Dentistry, Tokyo, Japan;
5. The Lion Foundation for Dental Health, Tokyo, Japan.
Received 2016-10-24; Accepted 2017-3-1; Published 2017-4-9
Abstract
The association between obesity and inflammation is well documented in epidemiological studies. Proteolysis of extracellular matrix (ECM) proteins is involved in adipose tissue enlargement, and matrix metalloproteinases (MMPs) collectively cleave all ECM proteins. Here, we examined the effects of C-reactive protein (CRP), an inflammatory biomarker, on the expression of MMPs and tissue inhibitors of metalloproteinases (TIMPs), which are natural inhibitors of MMPs, in adipocyte-differentiated 3T3-L1 cells. We analyzed the expression of Fcγ receptor (FcγR) IIb and FcγRIII, which are candidates for CRP receptors, and the effects of anti-CD16/CD32 antibodies, which can act as FcγRII and FcγRIII blockers on CRP-induced alteration of MMP and TIMP expression. Moreover, we examined the effects of CRP on the activation of mitogen-activated protein kinase (MAPK) signaling, which is involved in MMP and TIMP expression, in the presence or absence of anti-CD16/CD32 antibodies. Stimulation with CRP increased MMP-1, MMP-3, MMP-9, MMP-11, MMP-14, and TIMP-1 expression but did not affect MMP-2, TIMP-2, and TIMP-4 expression; TIMP-3 expression was not detected. Adipocyte-differentiated 3T3-L1cells expressed FcγRIIb and FcγRIII; this expression was upregulated on stimulation with CRP. Anti-CD16/CD32 antibodies inhibited CRP-induced expression of MMPs, except MMP-11, and TIMP-1. CRP induced the phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 and p38 MAPK but did not affect SAPK/JNK phosphorylation, and Anti-CD16/CD32 attenuated the CRP-induced phosphorylation of p38 MAPK, but not that of ERK1/2. These results suggest that CRP facilitates ECM turnover in adipose tissue by increasing the production of multiple MMPs and TIMP-1 in adipocytes. Moreover, FcγRIIb and FcγRIII are involved in the CRP-induced expression of MMPs and TIMP-1 and the CRP-induced phosphorylation of p38, whereas the FcγR-independent pathway may regulate the CRP-induced MMP-11 expression and the CRP-induced ERK1/2 phosphorylation.
Keywords: 3T3-L1 adipocyte, C-reactive protein, extracellular matrix, Fcγ receptor, matrix metalloproteinase, tissue inhibitor of metalloproteinase.
Introduction
Obesity is frequently associated with hyperglycemia, hyperinsulinemia, hypertension, and dyslipidemia [1, 2]; this cluster of metabolic disorders comprises metabolic syndrome, which is a known risk factor for cardiovascular disease [3,4] and type 2 diabetes [5,6]. Obesity onset and exacerbation arise from adipose tissue enlargement involving adipogenesis, angiogenesis, and proteolysis of extracellular matrix (ECM) proteins [7-10].
The matrix metalloproteinase (MMP) family comprises over 20 neutral endopeptidase that can collectively cleave all ECM and non-ECM proteins [11,12]. MMP activity depends on interactions between MMPs and tissue inhibitors of metalloproteinases (TIMPs), which are natural inhibitors of MMPs [11,12]. Changes in MMP and TIMP levels were observed in an obesity mouse model [13-15], suggesting that the MMP and TIMP system has a potential role in obesity development following adipose tissue hypertrophy and hyperplasia.
C-reactive protein (CRP) is the most extensively studied inflammatory biomarker. The association of elevated CRP levels with obesity, cardiovascular disease, and diabetes development has been well documented in epidemiological and in vivo experimental studies [16-21]. Fcγ receptors (FcγRs), a family of glycoproteins, bind to extracellular IgGs and to CRP or serum amyloid P, which are involved in the innate immune system [22,23]. Thus, CRP can act as an FcγR ligand [21,24]. In human cells, three FcγR classes have been identified: FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16) [22,23]. A study using human histiocytes indicated that CRP induced MMP-1 expression via FcγRII [25]. The effects of CRP on adipocytes acting as endocrine cells, secreting various adipokines, were reported by Yuan et al. [26,27]; their in vitro studies using 3T3-L1 murine adipocytes revealed that CRP suppresses adiponectin and leptin expression but induces interleukin (IL)-6 and tumor necrosis factor (TNF)-α expression. Less information is available about the effect of CRP on MMP and TIMP expression, and activation of mitogen-activated protein kinase (MAPK) signaling, which regulates MMP and TIMP expression [28-29], in adipocytes. Here, we focused on the degradation of ECM, which is involved in adipose tissue enlargement, and conducted an in vitro study to examine the effects of CRP on MMP and TIMP expression in adipocytes. We investigated the effects of anti-CD16/CD32 antibodies (Abs), which can act as blockers of FcγRs, on CRP-induced alteration of MMP and TIMP expression. Moreover, we examined the effect of CRP on the phosphorylation of extracellular signal-regulated kinase (ERK) 1/2, p38 MAPK, and stress-activated protein kinases/c-jun N-terminal kinases (SAPK/JNK) in the presence or absence of anti-CD16/CD32 Abs.
Material and methods
Cell culture and differentiation
We used cells of the mouse embryo cell line 3T3-L1 as model preadipocytes. 3T3-L1 (Riken BioResource Center, Tsukuba, Japan) cells were cultured at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL, Rockville, MD, USA) containing 25 mM glucose, 10% heat-inactivated fetal bovine serum (FBS; Gibco-BRL), and 1% (v/v) penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA). At confluence, 3T3-L1 cells were cultured for 2 days in DMEM further supplemented with 1 μM insulin, 0.5 μM isobutylmethylxanthine, and 0.1 μM dexamethasone (AdipoInducer Reagent; Takara Bio, Shiga, Japan). On day 2 and thereafter, DMEM containing 10% FBS, 1% (v/v) penicillin/streptomycin, and 1 μM insulin was subsequently replaced every 2 days. By day 8, 90% of the preadipocytes differentiated into adipocytes, as determined by lipid accumulation visualized with Oil Red O staining.
Stimulation with CRP
Adipocytes were starved for 6 h in FBS-free medium and then stimulated with 0, 25, or 50 μg/mL human recombinant CRP (Calbiochem, La Jolla, CA, USA) for 12 h. The CRP concentration range was chosen based on previous studies [26,27]. To investigate the role of FcγRs in CRP-induced alteration of MMP and TIMP expression in adipocytes, the cells were cultured in the presence or absence of 1.0 µg/mL anti-CD16/CD32 Abs (Abcam, Cambridge, MA, USA) for 1 h before stimulation with CRP. The Ab concentrations used were based on manufacturer instructions. CRP and anti-CD16/CD32 Abs did not apparently affect cellular lipid accumulation or architecture (Fig. 1).
Lipid accumulation in CRP-stimulated and unstimulated 3T3-L1 cells. Differentiated 3T3-L1 cells were cultured with 0 (control) or 50 µg/mL CRP in the presence or absence of anti-CD16/32 Abs for 12 h; cells were stained with Oil Red O.
Real-time reverse transcription (RT)-polymerase chain reaction (PCR)
Total RNA was isolated using NucleoSpin RNA (Takara Bio) and treated with DNase. mRNA was converted into complementary DNA (cDNA) with an RNA PCR kit (PrimeScript; Takara Bio). The resulting cDNA mixture was diluted 1:2 in sterile distilled water, and 2 µL diluted cDNA was subjected to real-time polymerase chain reaction (PCR) with SYBR Green I. The reactions were performed in 25 µL SYBR premixed Ex Taq solution (Takara Bio) containing 10 µM sense and antisense primers (Table 1). The PCRs were performed using a Thermal Cycler Dice Real Time System (Takara Bio) and analyzed using the instrument's software. The protocol for MMPs, TIMPs, and FcγRs was 40 cycles at 95°C for 5 s and 60°C for 30 s. All real-time PCR experiments were performed in triplicate; product specificity was verified through melting curve analysis. Calculated gene expression levels were normalized to 36B4 mRNA levels.
PCR primers used in the experiments.
| Target | Forward primer | Reverse primer | Genbank acc no. |
|---|---|---|---|
| MMP-1 | 5'-CACATTGATGCTGCTGTTGTGA-3' | 5'-TCTGCTGTTAATCTGGGATAACCTG-3' | NM_032006.3 |
| MMP-2 | 5'-GATAACCTGGATGCCGTCGTG-3' | 5'-GGTGTGCAGCGATGAAGATGATA-3' | NM_008610.2 |
| MMP-3 | 5'-CTCATGCCTATGCACCTGGAC-3' | 5'-TCATGAGCAGCAACCAGGAA-3' | NM_010809.1 |
| MMP-9 | 5'-GCCCTGGAACTCACACGACA-3' | 5'-TTGGAAACTCACACGCCAGAAG-3' | NM_013599 |
| MMP-11 | 5'-TGGAGACTATTGGCGTTTCCAC -3' | 5'-TTCACGGGATCAAACTTCCAG -3' | NM_008606 |
| MMP-13 | 5'-TCCCTGGAATTGGCAACAAAG-3' | 5'-GCATGACTCTCACAATGCGATTAC-3' | NM_008607.2 |
| MMP-14 | 5'-GCAGTGGACAGCGAGTACCCTA-3' | 5'-AGTCCCGCAGAGCTGACTTG-3' | NM_008608.3 |
| TIMP-1 | 5'-CTATAGTGCTGGCTGTGGGGTGTG-3' | 5'-TTCCGTGGCAGGCAAGCAAAGT-3' | NM_001044384.1 |
| TIMP-2 | 5'-GGCCTCCCTCCCTTACTCC-3' | 5'-GACTTCATATTCCAGCACGCACAT-3' | NM_011594.3 |
| TIMP-3 | 5'-CTAAGTCGGCTGTTTGGGTTGA-3' | 5'-CAGCACAGCTGGCTTGCTAGA-3' | NM_011595.2 |
| TIMP-4 | 5'-GCCTGAATCATCACTACCACCAGA-3' | 5'-TGAGATGGTACATGGCACTGCATA-3' | NM_080639.3 |
| FcγIIb | 5'-ACTTTGTGCCATATGCTACTGTGGA -3' | 5'-GAGTTTGACCACAGCCTTTGGAA -3' | NM_001077189 |
| FcγIII | 5'-GCCAATGGCTACTTCCACCAC -3' | 5'-GTCCAGTTTCACCACAGCCTTC -3' | NM_010188 |
| 36B4 | 5'- AAGCGCGTCCTGGCATTGTCT-3' | 5'-CCGCAGGGGCAGCAGTGGT -3' | NM_007475 |
SDS-PAGE and western blotting
Cells were lysed with extraction buffer containing 0.05% Triton X-100, 10 mM β-mercaptoethanol, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM ethylenediaminetetraacetic acid, and 25 mM Tris-HCl (pH 7.4). Cell membranes were disrupted by sonication, and the samples were clarified by centrifugation. Supernatants containing 20 µg intracellular protein were dissolved in 10 µL sample buffer containing 1% sodium dodecyl sulfate (SDS), 2 M urea, 15 mg/mL dithiothreitol, and bromophenol blue and heated at 95°C for 5 min before loading. The proteins were resolved by 4-20% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) with a discontinuous Tris-glycine buffer system [30], transferred to a polyvinylidene fluoride membrane by using a semidry transfer apparatus, and probed with Abs. The polyclonal or monoclonal IgG primary Abs used included the following: rabbit anti-MMP-2, anti-MMP-13, anti-TIMP-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-MMP-14 (Assay Biotech, Sunnyvale, CA, USA) Abs; goat anti-MMP-1, anti-MMP-3, anti-MMP-9, and anti-MMP-11 Abs (Santa Cruz Biotechnology); mouse anti-β-tubulin Abs (Santa Cruz Biotechnology); and rabbit anti-ERK1/2, anti-phospho-ERK1/2, anti-p38 MAPK, anti-phospho-p38 MAPK, anti-SAPK/JNK, and anti-phospho-SAPK/JNK (Cell Signaling Technology, Danvers, MA, USA) Abs. They were used with the appropriate biotin-conjugated donkey anti-goat IgG (Chemicon International, Temecula, CA, USA), goat anti-rabbit IgG (Zymed, San Francisco, CA, USA), or goat anti-mouse IgG (Abcam plc, Cambridge, UK) secondary Abs. The membranes were labeled with streptavidin-horseradish peroxidase (streptavidin-HRP) and visualized using a commercial chemiluminescence kit (Amersham Life Sciences, Little Chalfont, Buckinghamshire, UK). For reprobing with different Abs, the membrane was stripped with Restore PLUS Western blot stripping buffer (Thermo Scientific, Rockford, IL, USA) at room temperature for 15 min.
Statistical analysis
Values have been reported in terms of mean ± standard deviation (SD). Significant differences were determined using one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison test. Differences with p value <0.05 were considered statistically significant.
Results
Effect of CRP on MMP and TIMP mRNA expression
MMP and TIMP mRNA expression was determined by real-time PCR using 3T3-L1 cells cultured for 12 h with or without CRP. MMP-1, MMP-11, and MMP-13 expression significantly increased by 2-2.5, 1.5-3.0, and 2.0-2.6 fold, respectively, in cells stimulated with 25 and 50 μg/mL CRP, as compared to levels in unstimulated control cells (Fig. 2A, E, and F). MMP-2, MMP-3, MMP-9, and MMP-14 expression was significantly, i.e., 1.2, 1.8, 1.5, and 1.5-fold higher, respectively, in cells stimulated with 50 μg/mL CRP than in unstimulated control cells (Fig. 2B-D and G).
Effect of CRP on MMP and TIMP mRNA expression. Differentiated 3T3-L1 cells were cultured with 0 (control), 25, or 50 µg/mL CRP for 12 h and the mRNA expression of seven MMPs (A-G) and four TIMPs (H-J) was determined by real-time PCR. Each bar indicates the mean ± standard deviation (SD) of three independent experiments. *p < 0.05, **p < 0.01 (stimulation with CRP vs. control).
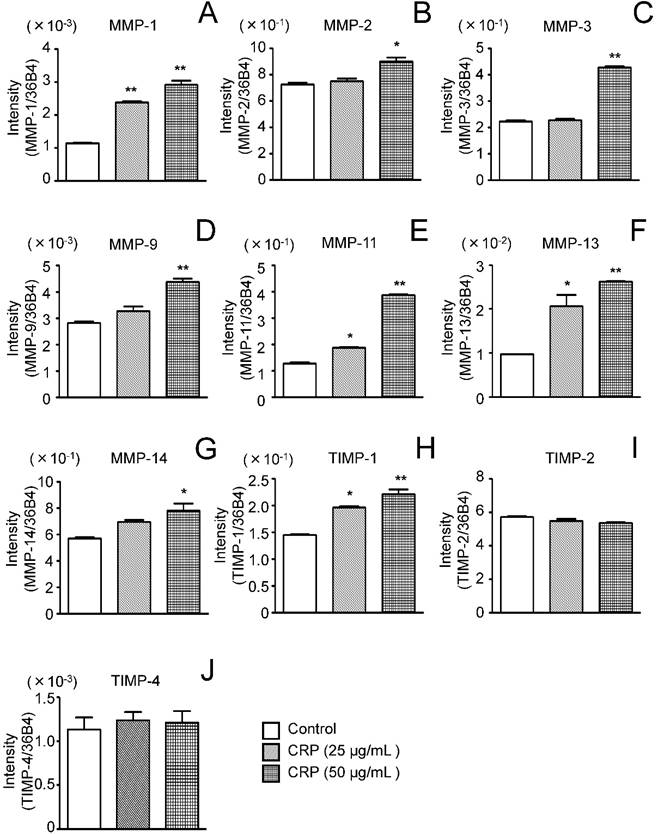
TIMP-1 mRNA expression was significantly, i.e., 1.3-1.5 fold, higher in cells stimulated with 25 or 50 μg/mL CRP than in unstimulated control cells, whereas TIMP-2 and TIMP-4 mRNA expression was unaffected by CRP (Fig. 2H-J). TIMP-3 mRNA expression was not detected in CRP-stimulated and unstimulated control cells (data not shown).
Effect of anti-CD16/CD32 Abs on CRP-induced MMP and TIMP-1 expression
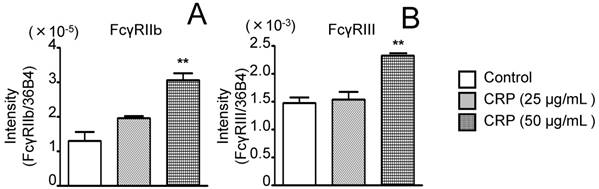
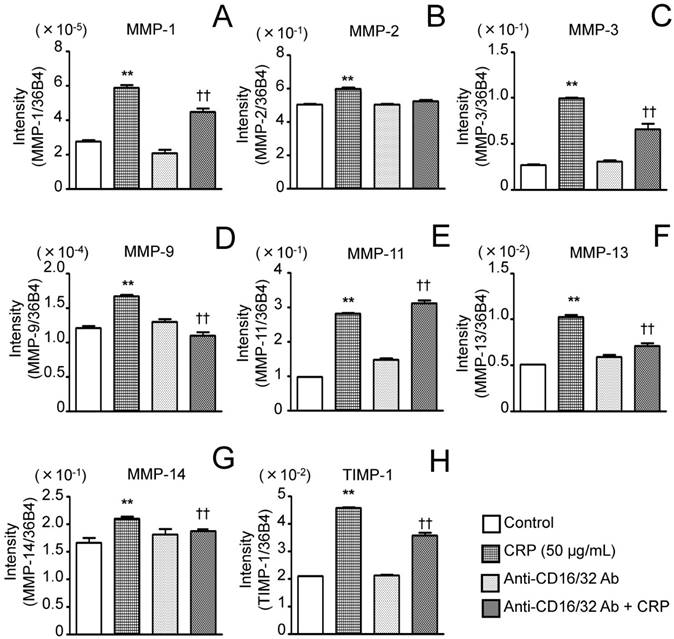
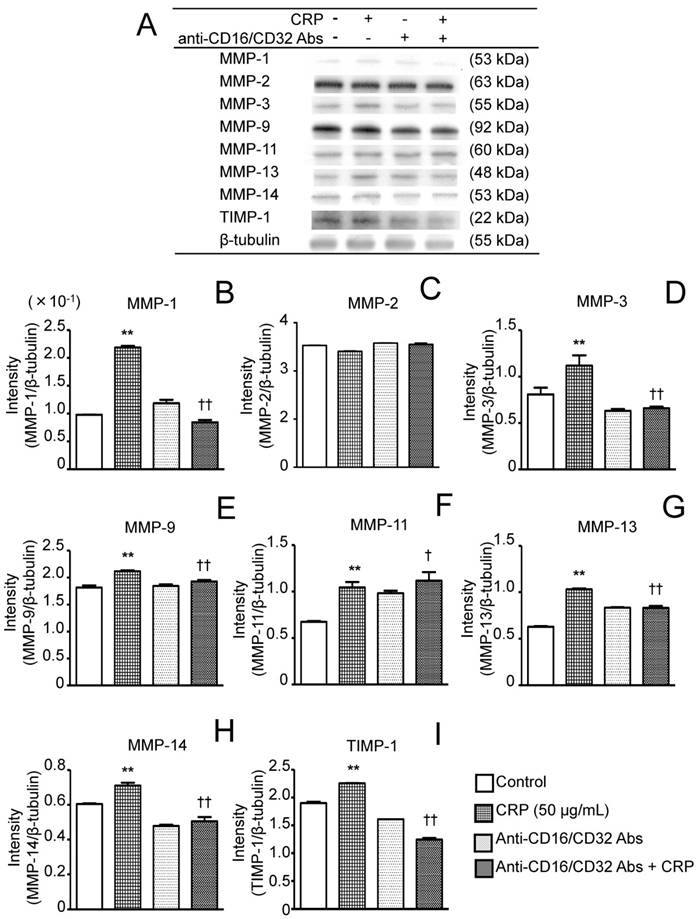
Before performing the inhibitory assay with anti-CD16/CD32 Abs, FcγRIIb (isoform of FcγRII) and FcγRIII mRNA expression in 3T3-L1 adipocytes was analyzed. CRP-stimulated and unstimulated control cells expressed FcγRIIb and FcγRIII mRNA; the expression of both receptors in the presence of 50 μg/mL CRP was significantly higher than that in the control (Fig. 3A and B). Next, 3T3-L1 adipocytes were stimulated with 50 μg/mL CRP in the presence or absence of anti-CD16/CD32 Abs, and MMP-1, MMP-2, MMP-3, MMP-9, MMP-11, MMP-13, MMP-14, and TIMP-1 expression was determined by real-time PCR and western blotting. MMP-1, MMP-2, MMP-3, MMP-9, MMP-13, MMP-14, and TIMP-1 mRNA expression in cells stimulated with CRP in the presence of Abs was significantly lower than that in cells stimulated with CRP in the absence of Abs (Fig. 4A-D, F-H). Thus, the anti-CD16/CD32 Abs blocked induction of MMPs and TIMP-1 mRNA expression by CRP. The effect of anti-CD16/CD32 Abs on CRP-induced MMP-11 expression was unexpected. MMP-11 expression was slightly higher in cells stimulated with CRP in the presence of Abs than in cells stimulated with CRP in the absence of Abs; this difference was statistically significant. Thus, anti-CD16/CD32 Abs slightly enhanced the effects of CRP on MMP-11 expression (Fig. 4E). The effects of stimulation with CRP on MMP and TIMP-1 protein expression in the presence or absence of anti-CD16/CD32 Abs were similar to those on mRNA expression, except in the case of MMP-2. CRP significantly induced MMP-1, MMP-3 MMP-9, MMP-13, MMP-14, and TIMP-1 protein expression, which was decreased by anti-CD16/CD32 Abs (Fig. 5A, B, D, E, G-I). MMP-11 protein expression was also increased by stimulation with CRP, and anti-CD16/CD32 Abs enhanced this increase (Fig. 5A and F). In contrast, MMP-2 protein expression levels in cells stimulated with CRP in both presence and absence of anti-CD16/CD32 Abs did not significantly differ from those in unstimulated control cells (Fig. 5A and C).
Effect of CRP on FcγRIIb and FcγRIII mRNA expression. Differentiated 3T3-L1 cells were cultured with 0 (control), 25, or 50 µg/mL CRP for 12 h; FcγRIIb (A) and FcγRIII (B) mRNA levels were determined by real-time PCR. Each bar indicates the mean ± standard deviation (SD) of three independent experiments. **p < 0.01 (stimulation with CRP vs. control)
Effect of anti-CD16/CD32 Abs on CRP-induced MMP and TIMP-1 mRNA expression. Differentiated 3T3-L1 cells were cultured with 0 (control) or 50 µg/mL CRP in the presence or absence of anti-CD16/CD32 Abs for 12 h; the mRNA expression of seven MMPs (A-G) and TIMP-1 (H) was determined by real-time PCR. Each bar indicates the mean ± standard deviation (SD) of three independent experiments. **p < 0.01 (stimulation with CRP vs. control), †p < 0.05, p < 0.01 (stimulation with CRP vs. anti-CD16/32 Ab + stimulation with CRP).
Effect of anti-CD16/CD32 Abs on CRP-induced MMP and TIMP-1 protein expression. Differentiated 3T3-L1 cells were cultured with 0 (control) or 50 µg/mL CRP in the presence or absence of anti-CD16/32 Abs for 12 h; the expression of seven MMPs and TIMP-1 protein was detected by western blotting (A). The blot intensities of MMPs (B-H) and TIMP-1 (I) were analyzed using a densitometer. Each bar indicates the mean ± standard deviation (SD) of three independent experiments. **p < 0.01 (stimulation with CRP vs. control), †p < 0.05, ††p < 0.01 (stimulation vs. anti-CD16/32 Ab + stimulation).
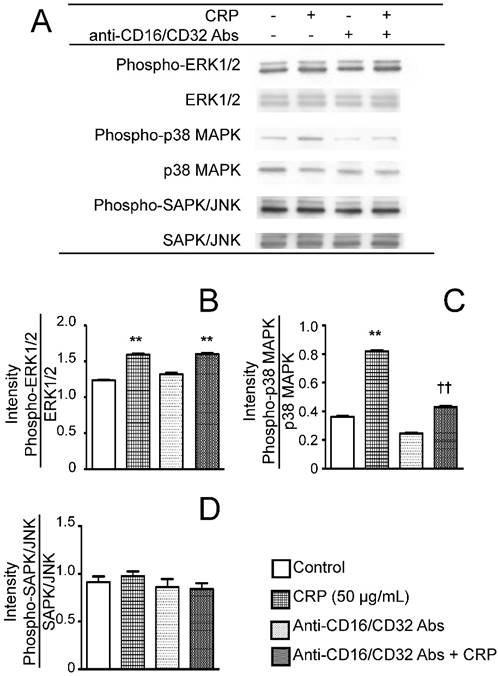
Effect of CRP and anti-CD16/CD32 Abs on the phosphorylation of ERK1/2, p38 MAPK, or SAPK/JNK
To determine the effects of CRP via FcγR on the activation of MAPK, the phosphorylation statuses of ERK1/2, p38 MAPK, or SAPK/JNK were determined after stimulating the cells with CRP in the presence or absence of anti-CD16/CD32 Abs. CRP induced the phosphorylation of ERK1/2 and p38 MAPK (Fig. 6A, B and C), but had no effect on SAPK/JNK phosphorylation (Fig. 6A and D). Anti-CD16/CD32 attenuated the CRP-induced phosphorylation of p38 MAPK, but not that of ERK1/2 (Fig. 6A, B, and C).
Effect of CRP and anti-CD16/CD32 on the phosphorylation of ERK1/2, p38 MAPK, and SAPK/JNK. Differentiated 3T3-L1 cells were cultured with 0 (control) or 50 µg/mL CRP in the presence or absence of anti-CD16/32 Abs for 12 h; the phosphorylation of ERK1/2, p38 MAPK, and SAPK/JNK was examined by western blotting (A). The blot intensities of the phosphorylated ERK1/2 (B), p38 MAPK (C), and SAPK/JNK (D) were analyzed using a densitometer. Each bar indicates the mean ± standard deviation (SD) of three independent experiments. **p < 0.01 (stimulation with CRP vs. control), ††p < 0.01 (stimulation vs. anti-CD16/32 Ab + stimulation).
Discussion
ECM proteins in adipose tissue undergo constant turnover, and MMPs are involved in the degradation of collagenous and noncollagenous proteins [7-12,31,32]. MMP-1 and MMP-13 (classified as collagenase-1 and collagenase-3, respectively) cleave the triple helix of fibrillar collagen (e.g., collagen I) into two fragments at the three-quarters position from the N terminus [11,12]. Subsequently, MMP-2 and MMP-9 (gelatinase-A and gelatinase-B, respectively) can degrade collagen fragments. In the current study, CRP induced MMP-1, MMP-13, and MMP-9 mRNA and protein expression [11,12]. CRP also upregulated MMP-2 mRNA but not protein expression. The increase in MMP-2 mRNA expression on stimulation with CRP (1.2 fold relative to control) was lower than that for other MMPs (1.5-3.0 fold relative to control); therefore, we considered that CRP had only a marginal effect on MMP-2 expression. These findings and our results indicated that CRP facilitated the overall process of fibrillar collagen degradation in adipose tissue via upregulation of collagenase (MMP-1 and MMP-13) and gelatinase (MMP-9) expression in 3T3-L1 adipocytes.
Mature adipocytes are surrounded by a basement membrane consisting of nonfibrillar collagen IV, laminin, and proteoglycans [8,9,31,33]. Collagen VI binds various other ECM components, including collagens I and IV [31,34]. Our in vitro study revealed that CRP induced MMP-3, MMP-11, and MMP-14 expression. MMP-3 and MMP-11 are classified as stromelysin-1 and stromelysin-3, respectively; the former degrades collagen IV, laminin, and proteoglycan, and the other mainly degrades collagen VI. MMP-14 (MT1-MMP) is expressed on the surface of cellular membranes and has broad substrate specificity [11,12]. Most MMPs are secreted as inactive proMMPs, whereas MMP-14 is expressed as an active enzyme on the cell surface and degrades ECM proteins, including collagen I, gelatin, laminin, and fibronectin, and activates proMMPs [11,35]. These findings and our results suggest that CRP facilitates proteolysis of both fibrillar and nonfibrillar ECM proteins in adipose tissue via stromelysin (MMP-3 and MMP-11) and MT-MMP (MMP-14) upregulation.
Posttranslational regulation of MMP activity depends on interactions between MMPs and TIMPs. TIMP-1 and TIMP-4 act as inhibitors against most types of MMPs, whereas they do not fully work as MMP-14 inhibitors; MMP-14 activity is suppressed by TIMP-2 and TIMP-3 [9,11,12]. Here, CRP increased TIMP-1 expression in adipocytes, whereas TIMP-2 and TIMP-4 expression was unaffected by stimulation with CRP. Degradation of ECM components in adipose tissue, including interstitial fibers and the basement membrane, allow adipocyte hypertrophy and hyperplasia in obesity [7-10]. The ECM acts as a frame maintaining the 3D structure of adipose tissue [9,31,33]. Thus, CRP might facilitate ECM degradation via upregulation of expression of multiple MMPs, whereas CRP-induced TIMP-1 might contribute to proteolysis regulation to maintain the frame of adipose tissue. Nutritional excess is one of the most important risk factors for obesity; association of obesity with systemic or local infection with organisms like Chlamydia pneumoniae or Porphyromonas gingivalis (periodontitis-related pathogen) has been documented [36-39]. These findings and our results suggest that elevation of CRP levels caused by these infections may act as a modification factor for exacerbating obesity via facilitation of ECM protein turnover in adipose tissue.
Here, we did not detect TIMP-3 mRNA expression in 3T3-L1 adipocytes. Bernot et al. [40] reported that mRNA expression of all four TIMPs in 3T3-L1 cells was markedly lower in adipocyte-differentiated 3T3-L1 than in undifferentiated cells. They found that TIMP-1, TIMP-2, and TIMP-4 expression remained at detectable levels during adipocyte conversion, whereas the decrease in TIMP-3 mRNA was substantial as compared to that of other TIMPs [40]. Here, we determined the mRNA expression levels of four TIMPs in adipocyte-differentiated 3T3-L1 cells; our result that TIMP-3 mRNA expression was not detected agreed with the findings of Bernot et al. [41].
FcγRIIa, one of the three FCγRII isoforms, has been identified as a CRP receptor in human immune cells [41,42]. Murine immune cells express FCγRIIb but not FCγRIIa and FCγRIIc [22,23]. Studies using mice reported that CRP induced insulin resistance thorough FCγRIIb [21,43]. This finding suggests that CRP can act as an FCγRIIb ligand in mice. Moreover, murine cells express FcγRIII, which is more closely related to human FCγIIa; these two receptors show high sequence homology in their extracellular domains [22,23]. To our knowledge, no study has reported FCγR expression in adipocytes. Here, we confirmed that adipocyte-differentiated 3T3-L1cells expressed both FCγRIIb and FcγRIII. Therefore, we focused on these two receptors and examined the effects of anti-CD16/CD32 Abs, which can act as FcγIII and FcγRII blockers, on CRP-induced MMP and TIMP-1 expression. Anti-CD16/CD34 Abs inhibited CRP-induced expression of MMPs, except MMP-11, and TIMP-1. These results suggested that CRP increased these MMPs and TIMP-1 via FcγRIIb and/or FcγIII in 3T3-L1 mouse adipocytes. Studies using adipocyte-differentiated 3T3-L1 cells reported that CRP altered the expression of adipokines such as adiponectin, leptin, or TNF-α via phosphatidylinositol-3 kinase, which is located in the downstream signaling pathway of FCγRs [26,27]. However, they did not focus on the effects of CRP on MMP and TIMPs expression and did not examine FCγR expression in adipocytes. Our findings, i.e., the involvement of FCγRs in CRP-induced MMPs and TIMP-1 expression, represent the first report of this phenomenon, although we did not determine which signaling pathways located directly downstream of FCγRIIb or FCγRIII were closely related to CRP-induced MMP and TIMP-1 expression. Montero et al. previously indicated that CRP induced MMP-1 and MMP-10 expression in human umbilical vein endothelial cells (HUVECs) and human endothelial cells; however, HUVECs did not express FcγRII (CD32) or FcγRIII (CD16) [44]. Here, the increase in MMP-11 expression in CRP-stimulated cells was higher in the presence than absence of anti-CD16/CD32 Abs. The findings of Montero et al. [44] and our results indicated that there is FcγR-independent induction of MMP expression. Anti-CD16/CD32 Abs inhibited CRP binding to FcγRII and FcγRIII; thus, CRP-induced MMP-11 expression via an FcγR-independent pathway might be facilitated.
MMP and TIMP expression is regulated by the MAPK pathway in many cell types, including fibroblast-like synoviocytes [28] and osteoblasts [29]. Here, CRP had a stimulatory effect on ERK1/2 and p38 MAPK phosphorylation and had no effect on SAPK/JNK phosphorylation. These results suggest that CRP-induced phosphorylation of ERK1/2 and p38 MAPK might be involved in CRP-induced MMP and TIMP-1 expression. In addition, anti-CD16/CD34 Abs attenuated CRP-induced p38 phosphorylation, but did not affect CRP-induced ERK1/2 phosphorylation. These results suggest that CRP induced p38 MAPK phosphorylation via FcγRIIb and/or FcγIII, whereas CRP-induced ERK1/2 phosphorylation might be mediated via other CRP receptors, and not via FcγR. Further research is required to clarify the receptor and its downstream pathway that regulate the effects of CRP on MMP expression and the MAPK signaling pathway in adipocytes.
In conclusion, our results suggest that CRP facilitates ECM turnover in adipose tissue by increasing the production of multiple MMPs and TIMP-1 in adipocytes. Moreover, FcγRIIb and FcγRIII are involved in CRP-induced expression of MMPs and TIMP-1, and CRP-induced phosphorylation of p38, whereas the FcγR-independent pathway may regulate CRP-induced MMP-11 and CRP-induced ERK1/2 phosphorylation.
Abbreviations
ECM: extracellular matrix; MMP: matrix metalloproteinases; CRP: C-reactive protein; Ab: antibody; TIMP: tissue inhibitors of metalloproteinase; FcγR: Fcγ receptor; IL: interleukin; TNF: tumor necrosis factor; MAPK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase; SAPK/JNK: stress-activated protein kinases/c-jun N-terminal kinase
Acknowledgements
This study was supported by Grants-in Aid for Scientific Research (C) (grant nos. 24592842 and 25462942) from the Japanese Society for the Promotion of Science; the Promotion and Mutual Aid Corporation for Private Schools of Japan; the Sato Fund and the Uemura Fund, the Nihon University School of Dentistry; a grant from the Dental Research Center, the Nihon University School of Dentistry; and a Nihon University Multidisciplinary Research Grant (2014-2015).
Authors' contributions
Kumiko Nakai: conceptualized and designed the study and experiments, performed the experiments, analyzed and interpreted the data, and wrote the manuscript.
Hideki Tanaka: designed the experiments, performed the experiments, interpreted the data, provided reagents and materials, and reviewed the manuscript.
Kazuhiro Yamanaka: performed the experiments, analyzed the data, and provided materials and reagents.
Yumi Takahashi: performed the experiments, analyzed the data, and provided materials and reagents.
Fumiko Murakami: designed the experiment, analyzed the data, and provided materials and reagents.
Rieko Matsuike: performed the experiments, analyzed the data.
Jumpei Sekino: performed the experiments, analyzed the data.
Natsuko Tanabe: provided materials and reagents, interpreted the data, and reviewed the manuscript.
Toyoko Morita: provided materials and reagents, interpreted the data, and reviewed the manuscript.
Yoji Yamazaki: interpreted the data and reviewed the manuscript.
Takayuki Kawato: conceptualized and designed the study and experiments, analyzed and interpreted the data, and wrote the manuscript.
Masao Maeno: Conceptualized and designed the study, provided reagents, materials, and equipment, interpreted the data, and reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kim SH, Després JP, Koh KK. Obesity and cardiovascular disease: friend or foe? Eur Heart J. 2015 [Epub ahead of print]
2. Diaz-Melean CM, Somers VK, Rodriguez-Escudero JP. et al. Mechanisms of adverse cardiometabolic consequences of obesity. Curr Atheroscler Rep. 2013;15:364
3. Isomaa B, Almgren P, Tuomi T. et al. Cardiovascular morbidity and mortality associated with the metabobic syndrome. Diabetes Care. 2001;24:683-9
4. Lakka HM, Laaksonen DE, Lakka TA. et al. The metabolic syndrome and total and cardiovascular disease mortality in middle-age men. JAMA. 2002;288:2709-16
5. Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. 2005;28:1769-78
6. Vasudevan AR, Ballantyne CM. Cardiometabolic risk assessment: an approach to the prevention of cardiovascular disease and diabetes mellitus. Clin Cornerstone. 2005;7:716
7. Christiaens V, Scroyen I, Lijnen HR. Role of proteolysis in development of murine adipose tissue. Thromb Haemost. 2008;99:290-4
8. Chun TH. Peri-adipocyte ECM remodeling in obesity and adipose tissue fibrosis. Adipocyte. 2012;1:89-95
9. Mariman EC, Wang P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol Life Sci. 2010;67:1277-92
10. Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094-101
11. Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562-73
12. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827-39
13. Lijnen HR, Maquoi E, Demeulemeester D. et al. Modulation of fibrinolytic and gelatinolytic activity during adipose tissue development in a mouse model of nutritionally induced obesity. Thromb Haemost. 2002;88:345-53
14. Maquoi E, Munaut C, Colige A. et al. Modulation of adipose tissue expression of murine matrix metalloproteinases and their tissue inhibitors with obesity. Diabetes. 2002;51:1093-101
15. Lijnen HR, Maquoi E, Holvoet P. et al. Adipose tissue expression of gelatinases in mouse models of obesity. Thromb Haemost. 2001;85:1111-6
16. Nguyen XM, Lane J, Smith BR. et al. Changes in inflammatory biomarkers across weight classes in a representative US population: a link between obesity and inflammation. J Gastrointest Surg. 2009;13:1205-12
17. Ishii S, Karlamangla AS, Bote M. et al. Obesity and repeated elevation of C-reactive protein: data from the CARDIA cohort. PLoS One. 2012;7:e36062
18. Lee S, Kim IT, Park HB. et al. High-sensitivity C-reactive protein can predict major adverse cardiovascular events in Korean patients with type 2 diabetes. J Korean Med Sci. 2011;26:1322-7
19. Festa A, D'Agostino R Jr, Tracy RP. et al. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. 2002;51:1131-7
20. Indulekha K, Surendar J, Mohan V. High sensitivity C-reactive protein, tumor necrosis factor-α, interleukin-6, and vascular cell adhesion molecule-1 levels in Asian Indians with metabolic syndrome and insulin resistance (CURES-105). J Diabetes Sci Technol. 2011;5:982-8
21. Tanigaki K, Vongpatanasin W, Barrera JA. et al. C-reactive protein causes insulin resistance in mice through Fcγ receptor IIB-mediated inhibition of skeletal muscle glucose delivery. Diabetes. 2013;62:721-31
22. Carlos R, Uribe-Querol E. Fc receptors: cell activators of antibody functions. Adv Biosci Biotechnol. 2013;4:21-33
23. Guilliams M, Bruhns P, Saeys Y. et al. The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol. 2014;14:94-108
24. Marnell L, Mold C, Du Clos TW. C-reactive protein: Ligands, receptors and role in inflammation. Clin Immunol. 2005;117:104-11
25. Williams TN, Zhang CX, Game BA. et al. C-reactive protein stimulates MMP-1 expression in U937 histiocytes through FcγRII and extracellular signal-regulated kinase pathway: an implication of CRP involvement in plaque destabilization. Arterioscler Thromb Vasc Biol. 2004;24:61-6
26. Yuan G, Chen X, Ma Q. et al. C-reactive protein inhibits adiponectin gene expression and secretion in 3T3-L1 adipocytes. J Endocrinol. 2007;194:275-81
27. Yuan G, Jia J, Di L. et al. Effects of C-reactive protein on adipokines genes expression in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2012;424:462-8
28. Wang P, Yang L, You X. et al. Mechanical stretch regulates the expression of matrix metalloproteinase in rheumatoid arthritis fibroblast-like synoviocytes. Connect Tissue Res. 2009;50:98-109
29. Nakai K, Kawato T, Morita T. et al. Angiotensin II induces the production of MMP-3 and MMP-13 through the MAPK signaling pathways via the AT1 receptor in osteoblasts. Biochimie. 2013;95:922-33
30. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680-5
31. Huang G, Greenspan DS. ECM roles in the function of metabolic tissues. Trends Endocrinol Metab. 2012;23:16-22
32. Zhou W, Liu G, Yang S. et al. Investigation for effects of cyclical dynamic compression on matrix metabolite and mechanical properties of chondrocytes cultured in alginate. J Hard Tissue Biol. 2016;25:351-6
33. Chun TH, Inoue M. 3-D adipocyte differentiation and peri-adipocyte collagen turnover. Methods Enzymol. 2014;538:15-34
34. Maraldi NM, Sabatelli P, Columbaro M. et al. Collagen VI myopathies: from the animalmodel to the clinical trial. Adv Enzyme Regul. 2009;49:197-211
35. Itoh Y, Seiki M. MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol. 2006;206:1-8
36. Rantala A, Lajunen T, Juvonen R. et al. Chlamydia pneumoniae infection is associated with elevated body mass index in young men. Epidemiol Infect. 2010;138:1267-73
37. Chitsazi MT, Pourabbas R, Shirmohammadi A. et al. Association of periodontal diseases with elevation of serum C-reactive protein and body mass index. J Dent Res Dent Clin Dent Prospects. 2008;2:9-14
38. Paraskevas S, Huizinga JD, Loos BG. A systematic review and meta-analyses on C-reactive protein in relation to periodontitis. J Clin Periodontol. 2008;35:277-90
39. Tsumori N, Kono T, Shigematsu N. et al. VEGF expression in diabetic rats promotes alveolar bone resorption by Porphyromonas gingivalis LPS. J Hard Tissue Biol. 2015;25:41-8
40. Bernot D, Barruet E, Poggi M. et al. Down-regulation of tissue inhibitor of metalloproteinase-3 (TIMP-3) expression is necessary for adipocyte differentiation. J Biol Chem. 2010;285:6508-14
41. Bharadwaj D, Stein MP, Volzer M. et al. The major receptor for C-reactive protein on leukocytes is Fcγ receptor II. J Exp Med. 1999;190:585-90
42. Raaz-Schrauder D, Ekici AB, Klinghammer L. et al. The proinflammatory effect of C-reactive protein on human endothelial cells depends on the FcγRIIa genotype. Thromb Res. 2014;133:426-32
43. Tanigaki K, Mineo C, Yuhanna IS. et al. C-reactive protein inhibits insulin activation of endothelial nitric oxide synthase via the immunoreceptor tyrosine-based inhibition motif of FcγRIIB and SHIP-1. Circ Res. 2009;104:1275-82
44. Montero I, Orbe J, Varo N. et al. C-reactive protein induces matrix metalloproteinase-1 and -10 in human endothelial cells: implications for clinical and subclinical atherosclerosis. J Am Coll Cardiol. 2006;47:1369-78
Author contact
![]() Corresponding author: Takayuki Kawato, DDS, PhD., Department of Oral Health Sciences, Nihon University School of Dentistry, 1-8-13, Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan Tel.: +81-3-3219-8128; Fax: +81-3-3219-8138. E-mail: kawato.takakyukiac.jp
Corresponding author: Takayuki Kawato, DDS, PhD., Department of Oral Health Sciences, Nihon University School of Dentistry, 1-8-13, Kanda Surugadai, Chiyoda-ku, Tokyo 101-8310, Japan Tel.: +81-3-3219-8128; Fax: +81-3-3219-8138. E-mail: kawato.takakyukiac.jp