Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(5):462-469. doi:10.7150/ijms.18154 This issue Cite
Research Paper
Induction of Mitotic Delay in Pharyngeal and Nasopharyngeal Carcinoma Cells Using an Aqueous Extract of Ajuga bracteosa
Yu-Tsai Lin 1,2 ![]() , Chih-Che Lin3, Hung-Chen Wang4, Yi-Chiang Hsu3,5
, Chih-Che Lin3, Hung-Chen Wang4, Yi-Chiang Hsu3,5 ![]()
1. Department of Otolaryngology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan;
2. Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, Taoyuan City, Taiwan;
3. Graduate Institute of Medical Sciences, College of Health Sciences, Chang Jung Christian University, Tainan, Taiwan;
4. Department of Neurosurgery, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan;
5. Bachelor Degree Program of Medical Sciences Industry, College of Health Sciences, Chang Jung Christian University, Tainan, Taiwan.
Received 2016-10-31; Accepted 2017-3-6; Published 2017-4-9
Abstract
Nasopharyngeal carcinoma (NPC) is a common cancer in Southeast Asia, for which radiotherapy and/or chemotherapy are the primary treatment methods. Many herbs are known to have potential uses in chemotherapy; however, the mechanisms underlying the observed antitumor activity of Ajuga bracteosa (AB) against NPC remain unclear. We explored the antitumor effects of AB, which was shown specifically to induce mitotic delay in pharyngeal (Detroit 562) and nasopharyngeal (Hone-1) cancer cells. Proliferation of cancer cells after exposure to aqueous extract of A. bracteosa (AEAB) was assessed using the MTT assay. DNA content and cell cycle arrest induction were analyzed using flow cytometry. The expression of checkpoint kinase 2 (CHK2), cell division control protein 2 (CDC2), and cyclin B1 was investigated using qRT-PCR and Western blot analysis. Results indicated the inhibition of cancer cell growth following exposure to AEAB. In addition, AEAB induced the accumulation of G2/M-phase cells in cancer cell through the disassociation of CDC2/cyclin B1 complex. Our findings suggested that, in addition to the known effects of AEAB in NPC prevention, it may have antitumor activities against NPC cells. In conclusion, AEAB inhibits the growth of and induces mitotic delay in cancer cells, supporting its use as an anticancer agent.
Keywords: Aqueous extract of Ajuga bracteosa, nasopharyngeal carcinoma, mitosis delay
Introduction
Nasopharyngeal carcinoma (NPC) in Southeast Asia, the incidence rates are as high as of 20-30 per 100,000 in men populations and 8 to 15 cases per 100,000 in women populations, respectively, especially among Chinese people [1], with the age of onset trending towards being earlier than for other tumors; accordingly, most patients fall within the range of 30-50 years [2]. Genetic predisposition, Epstein-Barr virus infection, and dietary and environmental factors are all believed to play an important role in the development of NPC [3]. Radiotherapy is the mainstay of treatment; combined with chemotherapy, but with severe toxic side effects and relatively low 5-year survival rate in the late-stage [4, 5]. Thus, the development of novel compounds for molecular targeting should be a priority in cancer research. Among the various treatment modalities, traditional medicines with demonstrable antitumor activity are preferred [6]. The mechanism of activation of CDC2-cyclin B has been investigated extensively for use in chemotherapy. CDC2 is phosphorylated at Thr-14 and Tyr-15 in the ATP-binding G-loop by Wee1 or Myt1 kinase [7]. The latter phosphorylation maintains the CDC2/cyclin B complex in an inactive state until the onset of M phase, when these sites are dephosphorylated by the phosphatase CDC25 [8].
The aerial part of Ajuga bracteosa Wall ex Benth, also known as Neelkanthi, which belongs to the Labiatae family, has been used as either as a food material or a traditional oriental medicine [9]. Bractin A, bractin B, and bractic acid, a long-chain polyhydroxy acid, were isolated from the whole plant of Ajuga bracteosa (AB) along with four known diterpenoids [10]. AB has been widely used in folk medicine in Asian countries against gout, hepatitis, pneumonia, rheumatism, various neuro-inflammatory disorders [11], and as an antiplasmodial agent [12, 13].
In this study, we explored whether the aqueous extract of Ajuga bracteosa (AEAB) contributes to the anti-proliferation and G2/M arrest of NPC (Hone-1 cells) and pharyngeal carcinoma (Detroit 562 cells), with the intention that a scientific basis for clinical treatment by Ajuga bracteosa for NPC therapy can be established.
Materials and Methods
Materials
AB was obtained from Rich Fountain International Corp (Taiwan). Dimethyl sulfoxide (DMSO) and 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Minimum essential medium Eagle (MEM), RPMI 1640, fetal bovine serum (FBS), phosphate-buffered saline (PBS), trypsin sodium, pyruvate and antibiotics were purchased from GIBCO-BRL (Grand Island, NY, USA). Molecular weight markers were purchased from Bio-Rad Laboratories, Inc. (Hercules, CA, USA) and polyvinylidene fluoride membranes (PVDF) were obtained from Merck Millipore (Billerica, MA, USA). All compounds and reagents were of the highest analytic grade available.
Ajuga bracteosa aqueous extract preparation
AB (dry weight 400 g) was powdered in a mill of knives and boiled with 200 ml distilled water at 100°C for 4 h. The total extract was concentrated under reduced pressure at 37℃ for 48 h to yield 1.5 g powder of AEAB after solvent evaporation. AEAB solutions with a concentration of 0, 1.56, 3.13, and 6.25 mg/mL were dissolved with distilled water as the working concentration for the experiment.
Cells
Two epithelial tumor cell lines, human pharyngeal carcinoma (Detroit 562 cells) and human nasopharyngeal carcinoma (Hone-1 cells), were obtained from cell bank of the National Health Research Institutes (Taiwan). Detroit 562 cells (human pharyngeal carcinoma) were maintained on culture dishes in 90% minimum essential medium Eagle with 2 mM L-glutamine and Earle's BSS adjusted to contain 1.5 g/L sodium bicarbonate, 0.1 mM non-essential amino acids, and 1 mM sodium pyruvate with 10% FBS. Hone-1 (human nasopharyngeal carcinoma) cells were cultured in RPMI 1640 supplemented with 10% FBS v/v. The cells were cultured under 5% CO2 at 37°C.
Cell proliferation assay
Cells were seeded at 5000 cells/well into a 96-well culture plate. The cells were exposed to 0, 1.56, 3.13, or 6.25 mg/mL AEAB for 24 to 72 h. Cells in each well were then treated with MTT (1 mg/mL) for at least 4 h. The reaction was stopped by adding DMSO and the optical density at 540 nm (OD540) was measured using a multi-well plate reader. Background absorbance of the medium in the absence of cells was subtracted. All samples were assayed at least in triplicate and the mean was calculated for each experiment. The results are expressed as a percentage of the control, which was considered to be 100%. All assay results are expressed as the mean ± SEM.
Apoptosis measurements
Cells were first seeded in six-well culture plates (Orange Scientific, Braine-l'Alleud, Belgium). After treatment with AEAB for 4 h, the cells were harvested and centrifuged; the cell pellet was then resuspended in and incubated with 1× Annexin-binding buffer [5 μL of Annexin V-FITC (BD Pharmingen, San Jose, CA, USA) and 1 μL of 100 μg/mL propidium iodide (PI) working solution] for 15 min at room temperature. After incubation, the stained cells were detected on a FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA) and analyzed using WinMDI 2.9 free software (Becton-Dickinson, San Jose, CA, USA).
Cell cycle analysis
We used the fluorescent nucleic acid dye PI to identify the proportion of cells in each of the three interphase stages of the cell cycle. The cells were treated with AEAB for 24 h and then harvested and fixed in 1 mL of cold 70% ethanol for at least 8 h at -20°C. DNA was stained using a PI/RNaseA solution, and the DNA content was detected using a FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA). Data were analyzed using WinMDI 2.9.
Mitotic index analysis
The mitotic index was assessed based on MPM-2 (anti-phospho-Ser/Thr-Pro) expression. After treatment with AEAB, cells were harvested and fixed in 70% ethanol overnight. Cells were then washed and suspended in 100 µL of IFA-Tx buffer (4% FCS, 150 nM NaCl, 10 nM HEPES, 0.1% sodium azide, and 0.1% Triton X-100) with MPM-2 antibody at room temperature for 1 h. The cells were washed and resuspended in IFA-Tx buffer with a rabbit anti-mouse FITC-conjugated secondary antibody (1:50 dilution; Serotec) for 1 h at room temperature in the dark. Finally, the cells were washed and resuspended in 500 µL of PBS with 20 µg/mL PI (Sigma-Aldrich) for 30 min in the dark. MPM-2 expression was analyzed using the FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA). Data were analyzed using WinMDI 2.9.
Western blot analysis
Proteins (50-75 µg) were separated through 10%-12% SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked with blocking buffer (Odyssey; Li-COR, Lincoln, NE, USA) overnight and incubated with anti-β-actin (Sigma-Aldrich), anti-phospho-cell division cycle 25 (p-cdc25c; sc-12354), anti-CDC2 (p34; sc-747), anti-cyclin B1 (sc-752), and CHK2 (SC-136251; Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies for 90-120 min. The blots were then washed and incubated with a secondary antibody (IRDy; Li-COR) at 1:20,000 dilution for 30-45 min. Next, the antigens were visualized on a near-infrared imaging system (Odyssey; LI-COR) and data were analyzed using the software Odyssey 2.1.
Co-immunoprecipitation (Co-IP)
Co-IP is an effective approach to quantify protein-protein interactions in cells. Briefly, after incubation at room temperature overnight, 500 mg of cellular proteins were labeled using anti-CDC2 (p34; sc-747). The protein-antibody immunoprecipitates were collected using protein A/G plus-agarose beads (SC-2003, Santa Cruz Biotechnology, CA, USA). Following the final wash, the samples were boiled and centrifuged to transform the agarose beads into pellets. Finally, we conducted Western blot analysis of cyclin B1 proteins in the supernatant. Antigens were visualized using a chemiluminescence detection kit (ECL; Amersham Corp., Arlington Heights, IL, USA) and data were analyzed using Odyssey 2.1 software.
RT-PCR
A reverse transcriptase system (Promega, Southampton, UK) was used to synthesize cDNA from 1 μg of total RNA. Between 2 and 4 μL of cDNA was used for PCR analysis. PCR (50 μL) reactions were performed using 100 ng of each primer and 1 unit of Dynazyme II (Flowgen, Lichfield, UK). Thermal cycling was conducted for 35 cycles as follows: 98°C for 10 s, 66°C for 30 s, and 72°C for 1 min, using a thermal cycler (Progene, Techne Ltd., Cambridge, UK). A final extension of 72°C was performed for 10 min at the end of the 35 cycles. The primers (Table 1) used for amplification of the target genes were checked against all other gene sequences for specificity. PCR reactions were analyzed on 1.5% agarose/TAE minigels and stained using 0.5 μg/mL ethidium bromide. Gels were visualized using an UV CCD camera system (Appligene, B and L Systems, Zoetermeer, Netherlands).
Primer sequences for RT-PCR and qRT-PCR.
| Gene | Primer Sequence | |
|---|---|---|
| RT-PCR | ||
| GAPDH | Forward | AGGTCGGAGTCAACGGATTTG |
| Reverse | GTGATGGCATGGACTGTGGT | |
| CDC2 | Forward | CCATACCATTGACTAACTATGGAAGAT |
| Reverse | GTCAGAAAGCTACATCTTCTTAATCTG | |
| Cyclin B | Forward | CCATTATTGATCGGTTCATGCAGA |
| Reverse | CTAGTGCAGAATTCAGCTGTGGTA | |
| qRT-PCR | ||
| GAPDH | Forward | GTCTCCTCTGACTTCAACAGCG |
| Reverse | ACCACCCTGTTGCTGTAGCCAA | |
| CDC2 | Forward | GGAAACCAGGAAGCCTAGCATC |
| Reverse | GGATGATTCAGTGCCATTTTGCC | |
| Cyclin B | Forward | GACCTGTGTCAGGCTTTCTCTG |
| Reverse | GGTATTTTGGTCTGACTGCTTGC | |
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed using approximately 200 ng of SYBR Green PCR MasterMix in an ABI 7300 system (Applied Biosystems, Foster City, CA, USA). A total of 40 cycles of PCR were conducted as follows: 95°C for 120 s, 60°C for 30 s, and 72°C for 30 s. The primers used for amplification of the human genes are listed in Tables 1. Samples from three plates were run in duplicate and the threshold suggested by the software was adopted for Ct calculations. To normalize readings, we used Ct values obtained at 18 s as internal controls for each run, and were thus able to calculate a delta Ct value for each gene. All protocols adopted for qRT-PCR were in accordance with the manufacturer's instructions.
Statistical analysis
All data are presented as the mean (± SEM) of at least three separate experiments. The t test or one-way ANOVA with a post hoc test was used for statistical analysis. A p value < 0.05 was indicates statistical significance.
Results
AEAB inhibits the survival and proliferation in Hone-1 and Detroit 562 cell
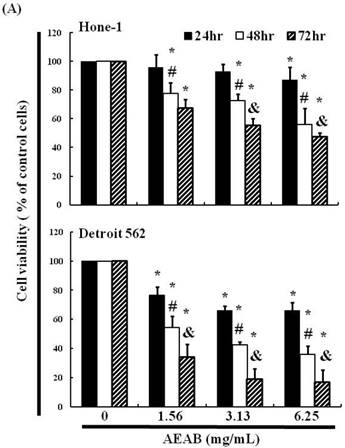
We hypothesized that AEAB may inhibit the survival of Hone-1 and Detroit 562 cells and thereby prevent proliferation. The antitumor activity of AEAB against NPC and pharyngeal cancer was investigated in vitro by treating Hone-1 and Detroit 562 cells with increasing doses of AEAB (0, 1.56, 3.13, and 6.25 mg/mL) for 24-72 h. We then measured the proliferation of AEAB-treated cancer cells using the MTT assay (Figure 1A). The findings indicated that the survival and proliferation of the Hone-1 and Detroit 562 cells decreased with an increasing AEAB dose. The same results were not obtained in CCD966SK (human normal fibroblasts) cells as control (normal cell) group (data not shown). Microscopic examination further revealed morphological changes in the cells following exposure to AEAB (3.13 mg/mL) for 6-24 h. AEAB also induced cancer cell death, as indicated by dead cells suspended in the medium (data not shown).
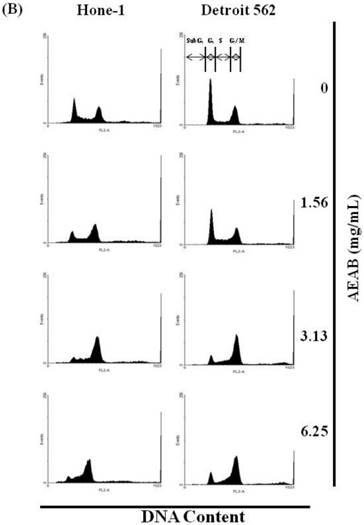
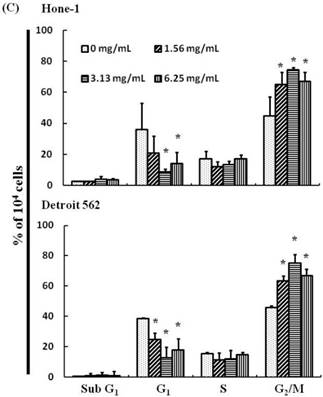
(A) ABAR inhibited proliferation of Hone-1 and Detroit 562 cells. Cells were treated with increasing doses of AEAB (0, 1.56, 3.13, and 6.25 mg/mL) for 24-72 h in vitro. The cell viability of AEAB-treated cancer cells was measured using the MTT assay. Results are expressed as a percentage of the control, which was set as 100%. All data are reported as the mean (± SEM) of at least three separate experiments. Statistical analysis was performed using the t-test, with differences between the treatment and control groups (0 mg/mL AEAB) considered significant at *P < 0.05 versus control group, #P < 0.05 versus 24 h-treated group and &P < 0.05 versus 48 h-treated group. (B) Effect of AEAB on cell cycle progression and distribution in Hone-1 and Detroit 562 cells. Cell cycle analysis of cancer cells by flow cytometry after being cultured with AEAB for 24 h. (C) The quantification results of cell cycle distribution were expressed as a percentage of G2/M-phase cell (%) treated with AEAB. Cells underwent staining with propidium iodide to analyze DNA content, which was then quantified through flow cytometry. An asterisk(*) in each group of bars indicates that the number of G2/M cells in the ABAR treatment group was significantly higher than that in the control group (P < 0.05).
Non-AEAB-induced apoptosis/necrosis in Hone-1 and Detroit 562 cell
To identify the role of AEAB in the apoptosis/necrosis of Hone-1 and Detroit 562 cell lines, we used Annexin V-FITC and PI staining to observe the formation of apoptotic cells following 4 h of exposure to AEAB. The percentage of apoptotic cells was assessed using flow cytometry. A dot-plot of Annexin V-FITC fluorescence versus PI fluorescence indicated a nonsignificant increase in the percentage of apoptotic cells upon treatment with AEAB compared with that in the untreated (control) cells (Figure S1A). Moreover, no significant increases were observed in the percentage of AEAB-treated Hone-1 and Detroit 562 cell lines, undergoing necrosis or apoptosis (Figure S1B).
AEAB-induced accumulation of G2/M-phase in Hone-1 and Detroit 562 cell
The cell cycle distribution of AEAB-treated cells was analyzed using flow cytometry. Cells were exposed to AEAB for 24 h before processing and analysis. As shown in Figure 1B, exposure to AEAB resulted in an increase in the number of cells in the G2/M phase, implying that the Hone-1 and Detroit 562 cells underwent a delay in mitosis. These results indicated that treatment with AEAB increased the number of cells in the G2/M phase while simultaneously reducing the number of cells in the G1 phase (Figure 1C).
Effects of AEAB on the mitotic index
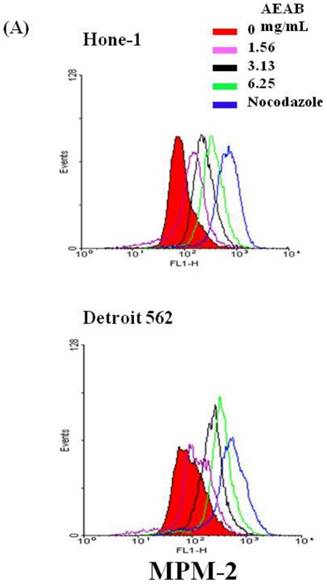
To differentiate G2 arrest from mitotic arrest, we used the marker MPM-2 (anti-phospho-Ser/Thr-Pro)-FITC, which can recognize proteins with epitopes exclusively phosphorylated during mitosis; specifically, from early prophase to metaphase. MPM-2 is commonly used as an indicator of mitotic disturbance. As a positive control, we treated separate groups of cells with nocodazole (15 µg/mL), an inducer of metaphase arrest, for 24 h, which resulted in synchronization of the entire cell population in the G2/M phase, as well as increased MPM-2 labeling (Figure 2A). MPM-2 levels in Hone-1 and Detroit 562 cells treated with AEAB were higher than those in the control group (Figure 2B). However, the effects of staining with MPM-2 were as strong as those of nocodazole in the group treated with 3.13 mg/mL AEAB, likely because cells stained with MPM-2 were in various stages of mitosis, some of which could not be identified using this early prophase marker. In particular, some G2/M-phase cells may have remained unstained. Thus, in addition to suggesting mitotic disturbance, increased MPM-2 staining may suggest its underestimation.
Diminished MPM-2 activity in AEAB-treated Hone-1 and Detroit 562 cells. MPM-2 activity of the cells was examined through flow cytometry 24 h after AEAB stimulation. (A) MPM-2 expression (gated cells) was performed using flow cytometry following treatment with AEAB for 24 h. (B) The quantification results were performed as a percentage of MPM-2 expression (%) in the cells treated with AEAB. As a positive control, separate groups of cells were treated for 24 h with nocodazole (15 µg/mL), an antifungal agent that induces metaphase arrest. An asterisk(*) in each group of bars indicates that the difference resulting from treatment with 0 mg/mL AEAB was statistically significant at P < 0.05.

G2/M-phase cell cycle arrest by AEAB in NPC cells through CDC2 and cyclin B1 disassociation
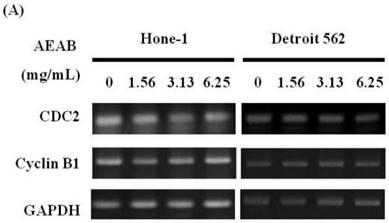
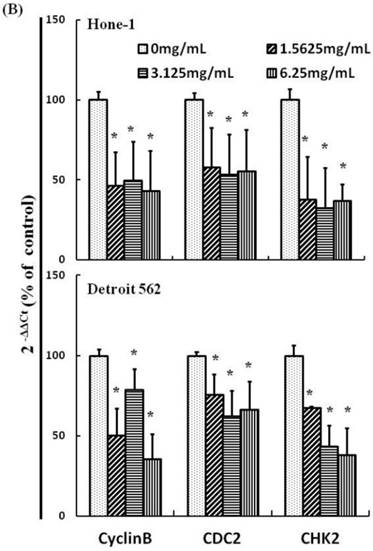
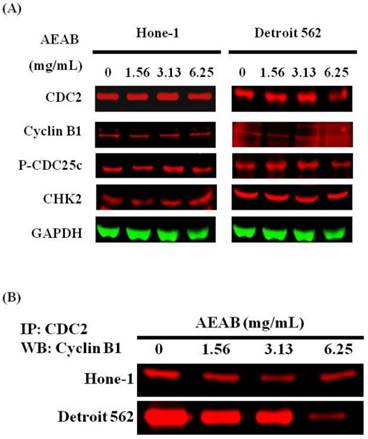
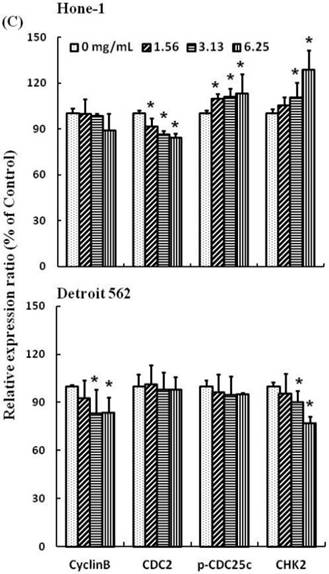
Figures 3A and 4A present the results of RT-PCR and Western blot analysis for cellular proteins from the Hone-1 and Detroit 562 cells treated with AEAB. In these analyses, CDC2 and cyclin B1 gene expression levels were quantified by measuring relative intensities. We observed no significant decrease in CDC2 and cyclin B1 mRNA levels following incubation with AEAB (Figure 3A). However, in qRT-PCR analysis, the gene levels of cyclin B1, CDC2 and CHK2 in AEAB treated-cell were significantly lower than in control (Figure 3B). Furthermore, the protein levels of cyclin B1 were significantly downregulated in Detroit 562 cells incubated with AEAB at concentrations of 3.13 to 6.25 mg/mL (Figure 4A and 4C). The protein expression of CDC2 in Hone-1 cells was also suppressed with AEAB treatment at concentrations of 1.56 to 6.25 mg/mL (Figure 4A and 4C). We also quantified the activity of the cyclin B1/CDC2 complex (crucial for G2-M transition during the cell cycle) by performing a Co-IP test (Figure 4B). The findings indicated that the increase in the number of NPC cells in the G2/M phase can be attributed to disassociation of the CDC2 and cyclin B1 complex following incubation with AEAB. The disassociation of the CDC2/cyclin B1 complex in NPC cell lines by AEAB may occur via the phosphorylation of CDC25c in Hone-1 cells and CHK2 downregulation in Detroit 562 cells (Figure 4C).
AEAB suppresses cyclin B, CDC2, and CHK2 gene expression in Hone-1 and Detroit 562 cells. Cells were treated with AEAB for 24 h. (A) The RT-PCR of cyclin B, CDC2, and CHK2 gene expression following exposure to the AEAB. (B) The panels indicate quantitative RT-PCR (qRT-PCR) analysis of cyclin B, CDC2, and CHK2 gene expression standardized against the level of GAPDH in cells. All data are reported as a percentage of 2-ΔΔCt at least three separate experiments (mean ± SEM). Statistical analysis was performed using the t-test, with differences considered significant at a level of *P < 0.05 versus the 0 mg/mL AEAB control group.
AEAB enhances CDC2/cyclin B1 disassociation in Hone-1 and Detroit 562 cells. Cells were treated with AEAB for 24 h, and protein expression was subsequently detected using (A) Western blot analysis, (B) Co-IP and (C) quantification of intensities for protein expression by Li-COR near infrared imaging system. All data are reported as the mean (± SEM) of at least three separate experiments. Statistical analysis was performed using the t test, with differences considered significant at a level of *P < 0.05 versus the 0 mg/mL AEAB control group.
Discussion
Our findings show that AEAB may irreversibly arrest Hone-1 and Detroit 562 cells growth. The results of our mechanistic analysis indicated that the inhibition of both cell proliferation and induction of cell mitosis delay are strongly dependent on AEAB accumulation in Hone-1 and Detroit 562 cells.
We observed that treating cancer cells with AEAB resulted in the downregulation of CDC2/cyclin B1 complex association, as well as CDC25c phosphorylation in Hone-1 cells. In addition, CDC25c was shown to bind to the CDC2/cyclin B1 complex, which mediates both the G2 and the M phases of the cell cycle [14]. The CDC25 families of proteins are highly conserved dual-specificity phosphatases that activate CDK complexes and respective cyclins, which in turn regulate cell cycle progression [15, 16]. During cell division, CDC25c dephosphorylates CDC2 to activate the CDC2/cyclin B1 complex, resulting in mitotic entry. Moreover, the CDC2/cyclin B1 complex phosphorylates CDC25c to enhance its phosphatase activity, leading to an irreversible auto-amplification loop into mitosis [17]. However, previous studies have shown that CDC25c inactivation promotes cancer development through mitotic delay [18, 19].
AEAB-induced delay in mitosis does not necessarily occur through the downregulation of cyclin B1 and CDC2 gene expression (Figures 3B and 4C), but rather through the upregulation of CHK2 in Hone-1 cells (Figure 4C).
Kayani WK, et al. indicated methanol extract of AB (AbMA) was found to be a promising bioactive extracts which had potent anti-oxidants, anti-inflammatory, analgesic, antidepressant and anticoagulant, were also good at in vivo activities. These activities can be attributed to the intrinsic polyphenols and phytoecdysteroids contents of AB [20]. Our results showed that AEAB may irreversibly arrest the growth of cancer cells and that both inhibition of proliferation and induction of mitosis delay are highly dependent on AEAB accumulation in cancer cells. We believe AEAB may have some potential bioactive components for antitumor activity, which remains unclear and will be investigated in our next work.
In conclusion, this is the first study demonstrating that Ajuga bracteosa is an effective inhibitor of NPC cell line. The role of AEAB in the inhibition of tumor growth was highlighted by the delay of mitosis through the disassociation of CDC2/cyclin B1 complex. These findings demonstrate the applicability of A. bracteosa as an anticancer agent for the chemoprevention of NPC. The effects of A. bracteosa and related compounds in cancer therapy and prevention should be explored. However, their potential as antitumor agents warrant further investigation.
Supplementary Material
Supplementary Figure S1A and Figure S1B.
Abbreviations
NPC: nasopharyngeal carcinoma; AB: Ajuga bracteosa; AEAB: aqueous extract of A. bracteosa; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; CHK2: checkpoint kinase 2; CDC2: cell division control protein 2; CDC25c: cell division cycle 25c; DMSO: dimethyl sulfoxide; MEM: minimum essential medium eagle; FBS: fetal bovine serum; PBS: phosphate-buffered saline; PVDF: polyvinylidene fluoride membranes; MPM-2: mitotic protein-2; OD540: optical density at 540 nm; Co-IP: co-immunoprecipitation; RT-PCR: reverse transcription polymerase chain reaction; qRT-PCR: quantitative real-time PCR; SEM: standard error of the mean.
Acknowledgements
The authors acknowledge funding received from Kaohsiung Chang Gung Memorial Hospital (Taiwan; grant NO.:CMRPG8E0271) and Chang Jung Christian University (Taiwan; grant NO.: Z099023).
Authorship
Yu-Tsai Lin and Yi-Chiang Hsu designed the experiments, analyzed the data and wrote the manuscript; Chih-Che Lin performed the experiments; Hung-Chen Wang analyzed the data. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. WHO. Union for International Cancer Control. Review of Cancer Medicines on the WHO List of Essential Medicines. Nasopharyngeal carcinoma. 2014
2. Chang MC, Chen JH, Liang JA. et al. Accuracy of whole-body FDG-PET and FDG-PET/CT in M staging of nasopharyngeal carcinoma: a systematic review and meta-analysis. Eur J Radiol. 2013;82:366-73
3. Su WH, Hildesheim A, Chang YS. Human leukocyte antigens and epstein-barr virus-associated nasopharyngeal carcinoma: old associations offer new clues into the role of immunity in infection-associated cancers. Front Oncol. 2013;3:299
4. Hao SP, Tsang NM. Surgical management of recurrent nasopharyngeal carcinoma. Chang Gung Med J. 2010;33:361-9
5. Fang FM, Tsai WL, Go SF. et al. Implications of quantitative tumor and nodal regression rates for nasopharyngeal carcinomas after 45 Gy of radiotherapy. Int J Radiat Oncol Biol Phys. 2001;50:961-9
6. Yu F, Tan WJ, Lu Y. et al. The other side of the coin: Leveraging Epstein-Barr virus in research and therapy. Oral Oncol. 2016;60:112-7
7. Jain P, Karthikeyan C, Moorthy NS. et al. Human CDC2-like kinase 1 (CLK1): a novel target for Alzheimer's disease. Curr Drug Targets. 2014;15:539-50
8. David M, Petit D, Bertoglio J. Cell cycle regulation of Rho signaling pathways. Cell Cycle. 2012;11:3003-10
9. Pal A, Toppo FA, Chaurasiya PK. et al. In-vitro cytotoxicity study of methanolic fraction from Ajuga Bracteosa wall ex. benth on MCF-7 breast adenocarcinoma and hep-2 larynx carcinoma cell lines. Pharmacognosy Res. 2014;6:87-91
10. Riaz N, Nawaz SA, Mukhtar N. et al. Isolation and enzyme-inhibition studies of the chemical constituents from Ajuga bracteosa. Chem Biodivers. 2007;4:72-83
11. Nisar A, Akhtar N, Hassan A. et al. Effect of Ajuga bracteosa on systemic T-cell immunity in Balb/C mice: dual Th1/Th2 immunostimulatory effects. Am J Chin Med. 2014;42:375-92
12. Chandel S, Bagai U. Antiplasmodial activity of Ajuga bracteosa against Plasmodium berghei infected BALB/c mice. Indian J Med Res. 2010;131:440-4
13. Chandel S, Bagai U. Screening of antiplasmodial efficacy of Ajuga bracteosa Wall ex. Benth. Parasitol Res. 2011;108:801-5
14. Liebermann DA, Tront JS, Sha X. et al. Gadd45 stress sensors in malignancy and leukemia. Crit Rev Oncog. 2011;16:129-40
15. Sur S, Agrawal DK. Phosphatases and kinases regulating CDC25 activity in the cell cycle: clinical implications of CDC25 overexpression and potential treatment strategies. Mol Cell Biochem. 2016;416:33-46
16. Brenner AK, Reikvam H, Lavecchia A. et al. Therapeutic targeting the cell division cycle 25 (CDC25) phosphatases in human acute myeloid leukemia-the possibility to target several kinases through inhibition of the various CDC25 isoforms. Molecules. 2014;19:18414-47
17. Lavecchia A, Di Giovanni C, Novellino E. Inhibitors of Cdc25 phosphatases as anticancer agents: a patent review. Expert Opin Ther Pat. 2010;20:405-25
18. Kiyokawa H, Ray D. In vivo roles of CDC25 phosphatases: biological insight into the anti-cancer therapeutic targets. Anticancer Agents Med Chem. 2008;8:832-6
19. Perona R, Moncho-Amor V, Machado-Pinilla R. et al. Role of CHK2 in cancer development. Clin Transl Oncol. 2008;10:538-42
20. Kayani WK, Dilshad E, Ahmed T. et al. Evaluation of Ajuga bracteosa for antioxidant, anti-inflammatory, analgesic, antidepressant and anticoagulant activities. BMC Complement Altern Med. 2016;16:375
Author contact
![]() Corresponding authors: Yi-Chiang Hsu, Graduate Institute of Medical Sciences, College of Health Sciences, Chang Jung Christian University, Tainan, 71101, Taiwan. E-mail: d8702008edu.tw or jenwaycjcu.edu.tw. Tel: +886-6-278-5123; Fax: +886-6-278-5663 Yu-Tsai Lin, Department of Otolaryngology, Kaohsiung Chang Gung Memorial Hospital, Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, 123 Ta-Pei Road, Niao-Song District, Kaohsiung, 83301, Taiwan E-mail: xeyeorg.tw or whc8131977com; Tel: +886-7-7317123 ext. 2533; Fax: +886-7-7313855
Corresponding authors: Yi-Chiang Hsu, Graduate Institute of Medical Sciences, College of Health Sciences, Chang Jung Christian University, Tainan, 71101, Taiwan. E-mail: d8702008edu.tw or jenwaycjcu.edu.tw. Tel: +886-6-278-5123; Fax: +886-6-278-5663 Yu-Tsai Lin, Department of Otolaryngology, Kaohsiung Chang Gung Memorial Hospital, Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, 123 Ta-Pei Road, Niao-Song District, Kaohsiung, 83301, Taiwan E-mail: xeyeorg.tw or whc8131977com; Tel: +886-7-7317123 ext. 2533; Fax: +886-7-7313855