Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(5):434-443. doi:10.7150/ijms.18427 This issue Cite
Research Paper
Dexamethasone Treatment at the Myoblast Stage Enhanced C2C12 Myocyte Differentiation
Der-Sheng Han1, 2, 3 ![]() , Wei-Shiung Yang4,5,6, Tung-Wei Kao7
, Wei-Shiung Yang4,5,6, Tung-Wei Kao7
1. Department of Physical Medicine and Rehabilitation
2. Community and Geriatric Medicine Research Center, National Taiwan University Hospital, BeiHu Branch, Taipei
3. Department of Physical Medicine and Rehabilitation
4. Department of Internal Medicine, National Taiwan University Hospital, Taipei
5. Graduate Institute of Clinical Medicine, College of Medicine, National Taiwan University, Taipei
6. Research Center for Developmental Biology and Regenerative Medicine, National Taiwan University, Taipei
7. Division of Geriatric Medicine, Department of Family and Community Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan.
Received 2016-11-20; Accepted 2017-3-1; Published 2017-4-8
Abstract
Background: Glucocorticoids induce skeletal muscle atrophy in many clinical situations; however, their hypertrophic and pro-differentiation effects on myotubes have rarely been reported. We hypothesized that dexamethasone (DEX) has a dual effect on muscle differentiation, and aimed to develop a new differentiation protocol for C2C12 cell line.
Methods: Dose- and time-dependent effect of DEX on C2C12 myoblast cell line was analyzed at myoblast and myotube stage, respectively. The level of differentiation was determined by myh1, pax7, atrogin-1, and myostatin mRNA expression and fusion index.
Results: After differentiation and at the myotube stage, DEX treatment has an atrophic effect. Specifically, the myotube was thinner, the expression of atrogin-1 increased, and the protein content of myosin heavy chain decreased. In contrast, when DEX treatment was performed before the onset of differentiation, we observed an increase in myotube diameter and myosin heavy chain levels, and a decrease in the expression of atrogin-1. The ratio of multinuclear myotube cells increased in the DEX treatment group. The optimal treatment concentration and time was 100 μM and 48 h, respectively. Co-treatment with 10 μM DEX and 100 nM insulin further enhanced the process of myotube differentiation.
Discussion: This novel finding contributed to the explanation on the stage-specific mechanism of glucocorticoid-induced myopathy. A new formula for myoblast differentiation, containing both DEX and insulin, is proposed. Further research is required to understand the complete mechanism of DEX-induced muscle hypertrophy.
Keywords: dexamethasone, glucocorticoid, muscle differentiation, myostatin, atrogin-1.
Introduction
Glucocorticoids have diverse and tissue-specific effects on cellular metabolism; they are generally believed to induce skeletal muscle atrophy. Steroid myopathy, first described by Cushing in 1932, is a disorder caused by excess steroids, either endogenous or exogenous. This condition results in varying degrees of muscle atrophy and proximal limb weakness [1]. There is evidence showing that fluorinated steroids such as dexamethasone (DEX) or triamcinolone, might frequently be a cause of this [2]. Glucocorticoids decrease protein synthesis and increase the rate of protein catabolism, thus leading to muscle atrophy [3]. Increased catabolism could result from diverse mechanisms, including inhibition of insulin and insulin-like growth factor I (IGF1), inhibition of protein synthesis machinery such as PI3K and mTOR, inhibition of satellite cell differentiation, and induction of myostatin, cathepsin, calpain, and the ubiquitin-proteosome system. Blockage of these catabolic pathways might reverse the process of muscle atrophy [4].
Muscle biologists frequently employ the differentiated C2C12 myoblast line as a cell model for mature myocytes. However, the differentiation process of C2C12 is tedious and difficult to learn. Skeletal muscle differentiation is a multi-stage highly-regulated process that includes myoblast division, elongation, and fusion [5]. During the process of C2C12 myoblast differentiation, we discovered that steroids have a distinct and opposite effect on myoblast differentiation from previous opinion, depending when they are applied, either before or after the onset of differentiation. In this study, we hypothesized that DEX has a dual effect in muscle differentiation, and aimed to morphologically, histochemically, and immunohistochemically analyze the dose- and time-dependent relationship with corticosteroids. In particular, we intended to determine the optimal treatment conditions of DEX for myoblast differentiation.
Materials and Methods
Cell culture
The C3H murine myoblast cell line C2C12 (American Type Culture Collection, Rockville, MD, USA) was grown in growth medium (GM), containing high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10 % fetal bovine serum, 100 U/ml of penicillin, and 100 μg/ml of streptomycin in a 5 % CO2 humidified atmosphere at 37 °C. Cells in T75 culture flasks were trypsinized and seeded at a density of 104 cells/cm2 in 12-well culture plates. For myotube experiments, in which differentiation of myoblasts into myotubes was induced, the medium was replaced with differentiation medium (DM), which was composed of DMEM containing antibiotics and 2 % horse serum, 72 h after seeding. The medium was changed every 48 h. After a 96-h incubation with differentiation medium, cells were treated with different concentrations of dexamethasone (DEX) for 48 h. The control group was treated with solvent only. For myoblast experiments, myoblasts were treated with growth medium containing different concentrations of DEX, for different incubation times after seeding. The medium was then changed to differentiation medium containing 100 nM insulin (Eli Lilly, Indianapolis, IN, USA) and/or 10 μM DEX every day for 96 h. Medium and serum were from Invitrogen (Grand Island, NY, USA). Antibiotics and trypsin were from Biological Industries (Israel). Drugs were from Sigma-Aldrich (St. Louis, MO, USA).
Measurement of myotube diameter
Myotube cultures were photographed with a phase contrast microscope (Leica DM IL, Germany) at 100 × magnification before extraction of RNA or proteins. The diameters of 100 myotubes were measured from 10 random fields by Image J software (NIH, Frederick, MD, USA) [6].
Quantification of RNA expression by quantitative real-time polymerase chain reaction (qPCR)
Myosin heavy chain (myh1), pax7, atrogin-1, and myostatin mRNA levels were quantified by real-time PCR. Primers and probes for myh1, pax7, atrogin-1, myostatin, and the housekeeping gene 36b4 were designed by Universal ProbeLibrary Assay Design Center (Roche Applied Science, Mannheim, Germany) [7]. 36b4 mRNA was the endogenous control employed to normalize mRNA concentrations. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Quantity and quality of the RNA extracts were measured using a Nanodrop (Thermo Scientific, Wilmington, DE, USA). First-strand cDNA was synthesized in a 20 μl reverse transcription (RT) reaction with 0.5 μg RNA using the QuantiTect reverse transcription kit (Qiagen, Venlo, Netherlands). The real-time PCR reaction was performed using a LightCycler® TaqMan® Master HybProbe kit (Roche Applied Science, Mannheim, Germany). The thermal amplification was conducted by a LightCycler® 2.0 Instrument (Roche Applied Science, Mannheim, Germany) according to the protocol provided by the manufacturer. The sequences of the forward and reverse primers, and TaqMan oligonucleotide probes (respectively) are listed below. myh1: 5′-GCC CAG TGG AGG ACA AAA TA-3′, 5′-TCT ACG TGC TCC TCA GCA T-3′, and 5′-CAT CCA GC-3′; pax7: 5′-GGC ACA GAG GAC CAA GCT C-3′, 5′-GCA CGC CGG TTA CTG AAC-3′, and 5′-TCC AGG TC-3′; atrogin-1: 5′-AGT GAG GAC CGG CTA CTG TG-3′, 5′-GAT CAA ACG CTT GCG AAT CT-3′, and 5′-CTC TGC CA-3′; myostatin: 5′-TGG CCA TGA TCT TGC TGT AA-3′, 5′-CCT TGA CTT CTA AAA AGG GAT TCA-3′, and 5′-CAG GAG AA-3′. Messenger RNA levels in control were arbitrarily set to 1.0.
Western Blotting
Western blotting was performed to determine protein levels of myosin and atrogin-1 in myotubes. Cells were lysed with RIPA buffer (1 % SDS, 150 mM NaCl, 1 % Triton X-100, 1 % sodium deoxycholate, and 20 mM Tris-HCl (J. T. Baker, Center Valley, PA, USA), pH 7.5) containing protease inhibitors (Roche Applied Science, Mannheim, Germany) and phosphatase inhibitors (Roche Applied Science, Mannheim, Germany), and centrifuged at 14,000 × g at 4 °C for 10 min. The supernatants were collected and the protein concentration was measured by the Micro BCA Protein assay reagent (Pierce, Rockford, IL, USA). Aliquots containing 20 μg of protein were diluted in 5 × loading buffer (10 % SDS, 50 % glycerol, 0.25 % bromophenol blue, and 0.25 M Tris-HCl, pH 6.8) and reduced at 37 °C for 15 min. Proteins were separated by 6 or 10 % sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis with 70 V for 30 min and then 110 V for 120 min using the Bio-Rad Mini-Protean II system (Bio-Rad, Hercules, CA, USA). The proteins were then transferred at 100 mA overnight onto nitrocellulose membranes (Amersham Bioscience, Buckinghamshire, UK), and blocked with TBST buffer (150 mM NaCl, 0.05 % Tween-20, and 20 mM Tris-HCl, pH 7.4) containing 5 % non-fatty milk (Anchor, Auckland, New Zealand) for 1 h at room temperature. After blocking, the membranes were incubated overnight with a 1:1000 dilution of primary antibody, which included mouse monoclonal antibody to myosin (MF20, R&D Systems, Minneapolis, MN, USA), rabbit monoclonal antibody to atrogin-1 (Abcam, Cambridge, MA, USA), and rabbit monoclonal antibody to α-tubulin (Cell signal, Beverly, MA, USA). The membranes were then washed and incubated for 1 h at room temperature with goat anti-rabbit or goat anti-mouse horseradish peroxidase-conjugated secondary antibody (GenTex, Rancho Cucamonga, CA, USA) at a 1:10,000 dilution. Following three washes with TBST, immunoreactive protein bands were visualized using Western LightningTM Plus-ECL oxidizing reagent plus (PerkinElmer, Waltham, MA, USA). Band intensity was analyzed by Multi Gauge software (Fujifilm, Sendai, Japan). The identity of the bands on the western blots was confirmed by comparing to the precision marker molecular weight marker (Thermo Scientific, Wilmington, DE, USA).
Immunocytochemistry
Sterile glass coverslips were coated with 10 % fetal bovine serum in a 6-well plate for 24 h. C2C12 cells were seeded on glass coverslips at a density of 2 × 104 cells/cm2 in GM. After treatment and culture, cells were fixed in 3 % paraformaldehyde in phosphate-buffered saline (Bioshop, Burlington, Canada) twice for 10 min, and then washed with PBS twice for 5 min. Fixed cells were permeabilized and blocked with 1 % bovine serum albumin (Bioshop) in PBST (PBS with 0.25 % Triton X-100) for 30 min at room temperature. Cells were then incubated with specific primary antibodies at a dilution of 1:100 in PBST with 1 % BSA, overnight at 4 °C. Cells were then washed three times for 5 min with PBST and incubated with secondary antibodies (Jackson Immuno Research, West Grove, PA, USA) at a dilution of 1:100 for 1 h at room temperature in PBST with 1% BSA. After washing with PBST three times, cells were incubated with 0.1 μg/ml DAPI (Jackson Immuno Research) for 5 min. After washing three times with PBST, coverslips were mounted with mounting medium (Abcam, Cambridge, MA, USA) and sealed with nail polish. Cells were observed with a fluorescence microscope (photo MicroGraph digitized integration system, Carl Zeiss, Germany) and then stored in the dark at -20 °C.
Statistical analysis
Results are presented as the means ± SEM. An independent t-test was employed to compare the mean cell diameter between different treatment groups. The Mann-Whitney U test was used to compare means in real time PCR and Western blotting analysis. Statistical analysis was performed using SPSS software, version 16.0 (Chicago, IL). A P-value < 0.05 was considered statistically significant.
Results
Dexamethasone has an opposite effect on differentiated and undifferentiated C2C12 myoblasts
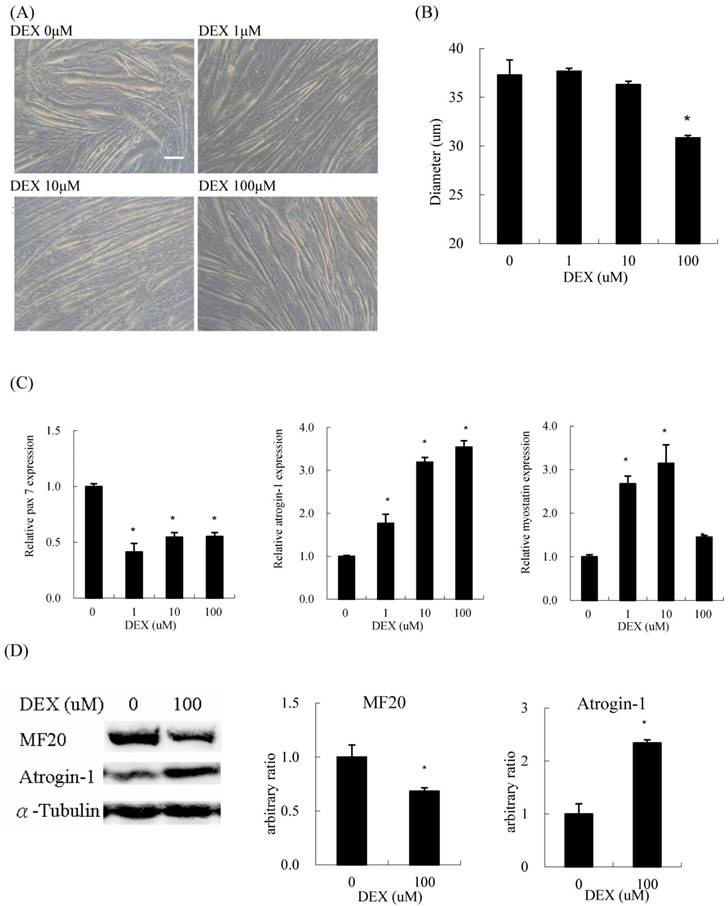
When differentiated C2C12 myoblasts were treated with DEX for 48 h, they became thinner compared to those without DEX treatment (Figure 1A). Regarding the concentration of DEX, 100 μM had the most prominent atrophic effect on myotube diameter among the test concentrations (Figure 1B). The expression of pax7, a myoblast marker, also decreased at a treatment concentration of 100 μM. The expression of atrogin-1, a catabolic (atrophic) marker, was induced by DEX in a dose-dependent manner. During treatment, the expression of myostatin, a negative feedback myokine controlling muscle mass, also increased (Figure 1C). The protein content of myosin heavy chain (MF20), a myofiber terminal differentiation marker, decreased, and that of atrogin-1 increased in myotubes treated with 100 μM DEX (Figure 1D). Evidence suggested that DEX has an atrophic effect when C2C12 cells are treated at the myotube stage.
Dexamethasone (DEX) treatment after differentiation induced myotube atrophy (A) Phenotypic change of C2C12 myotubes treated with 0, 1, 10, and 100 μM DEX after differentiation. Scale bar: 50 μm. (B) The diameter of myotubes treated with 100 μM DEX decreased significantly. (C) The mRNA expression level of myh1 and myostatin in myotubes treated with 1 and 10 μM DEX increased, but declined in myotubes treated with 100 μM DEX. The mRNA expression level of pax7 decreased and atrogin-1 increased in myotubes treated with DEX. (D) The protein expression level of myosin heavy chain (MF20) decreased and atrogin-1 increased in myotubes treated with 100 μM DEX. Results are presented as the mean ± SEM from three independent experiments. Asterisk indicates significant difference versus control (*p < 0.05).
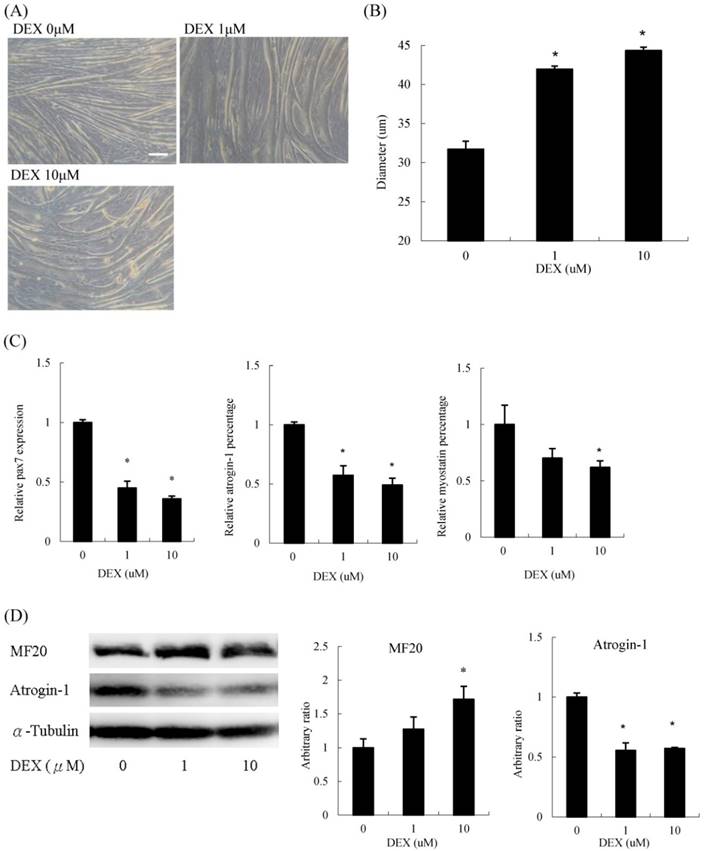
Interestingly, when we treated undifferentiated C2C12 myoblasts with DEX transiently for 48 h in GM, differentiation was significantly enhanced. The myotube diameter did not diminish but instead increased (Figure 2A). Myoblasts treated with 10 μM DEX had a 33 % increase in diameter compared to that of untreated myoblasts (Figure 2B). The mRNA expression of pax7 and atrophic molecular markers, including atrogin-1 and myostatin, decreased (Figure 2C). Regarding protein levels, myosin heavy chain levels increased, whereas atrogin-1 decreased upon DEX treatment (Figure 2D). Cells treated with 100 μM DEX at the undifferentiated stage could not be analyzed since some cells detached from the culture dishes during the differentiation process. Thus, DEX has a hypertrophic effect when C2C12 cells are pretreated at the myoblast stage.
Dexamethasone treatment before differentiation induced myotube hypertrophy (A) Phenotypic change in myotubes. Scale bar: 50 μm. (B) The diameter of myotubes increased. (C) The mRNA expression level of pax7, atrogin-1 and myostatin decreased. (D) The protein expression level of myosin heavy chain (MF20) increased and atrogin-1 decreased. Results are presented as the mean ± SEM from three independent experiments. Asterisk indicates significant difference versus control (*p < 0.05).
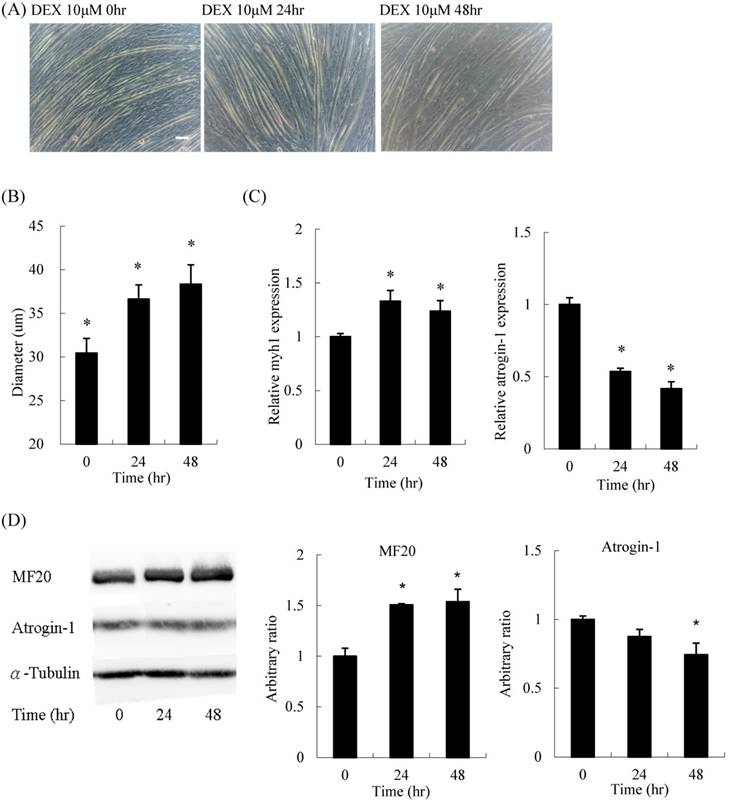
For the time-course experiment, myoblasts pretreated with DEX for 48 h had the thickest diameter (Figures 3A and B). Both mRNA expression and protein content of myh1 were increased, whereas atrogin-1 was significantly decreased after DEX treatment for 48 h (Figures 3C and D). Thus, the optimal treatment duration was 48 h.
Time course effect of 10 μM DEX treatment on myoblasts before differentiation (A) Phenotypic change in myotubes. Scale bar: 50 μm. (B) The diameter of myotubes increased after treatment for 24 and 48 h. (C) The mRNA expression level of atrogin-1 decreased after treatment for 24 and 48 h. (D) Protein expression of myosin (MF20) increased and atrogin-1 decreased. Results are presented as the mean ± SEM from three independent experiments. Asterisk indicates significant difference versus control (*p < 0.05).
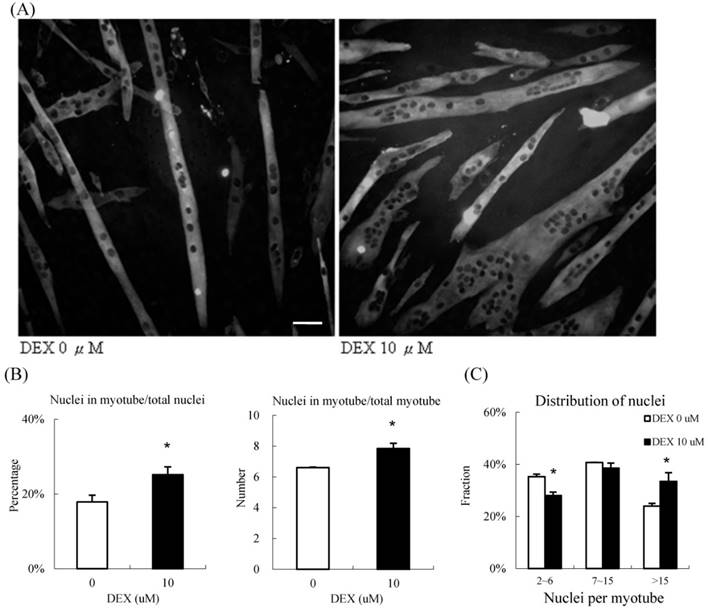
During the process of differentiation, mononuclear myoblasts will fuse to form the myotubes, which are large multinucleated cells. We calculated the fusion index, which is the nuclei distribution, to determine the extent of myotube differentiation, by immunocytochemisty (Figure 4A). In the group treated with 10 μM DEX, approximately 25 % of all nuclei in the high power field were within the myotubes; only 18 % of nuclei were within the myotubes of the control group. On average, there were 8.0 and 6.4 nuclei in each myotube in the treatment and control groups, respectively (Figure 4B). We further categorized the myotubes into three groups based on the number of nuclei, 2 to 6 nuclei per myotube (low), 7 to 15 nuclei per myotube (moderate), and > 15 nuclei per myotube (high)[8]. In the treatment group, nearly 40 % of nuclei were located in highly multinucleated myotubes; in contrast, only 28 % of nuclei were present in this condition in the control group. The distribution of nucleation shifted when we treated them with DEX (Fig 4C). Taken together, DEX treatment at the myoblast stage promoted myogenic differentiation and fusion index.
10 μM DEX treatment enhanced nuclei fusion in undifferentiated myoblasts (A) Representative images of undifferentiated myoblasts treated with 10 μM DEX. Myosin was labeled with green fluorescence, and the nuclei were labeled with DAPI (black dots inside myoblast). Scale bar: 50 μm. (B) The fusion index was quantified by the percentage of nuclei in myotubes, and the nuclei per myotube. The percentage of nuclei in myotubes and the nuclei per myotube increased when treated with 10 μM DEX. Myoblasts treated with 10 μM DEX had a high fusion index. (C) Myotubes were further categorized into three groups (2 to 6 nuclei, 7 to 15 nuclei, and >15 nuclei per myotube). The distribution of nuclei, and number of multinuclear myotubes in the three groups were calculated. Compared with that in the vehicle treatment group, the fraction of nuclei decreased in the 2 to 6 nuclei group but increased in > 15 nuclei group in myotubes treated with 10 μM DEX. The distribution of myotubes was not significantly different between controls and myotubes treated with 10 μM DEX. Results are presented as the mean ± SEM from three independent experiments. Asterisk indicates significant difference versus control (*p < 0.05). White bars indicate vehicle treatment; black bars indicate 10 μM DEX treatment.
DEX plus insulin treatment at the myoblast stage synergistically enhanced myogenic differentiation
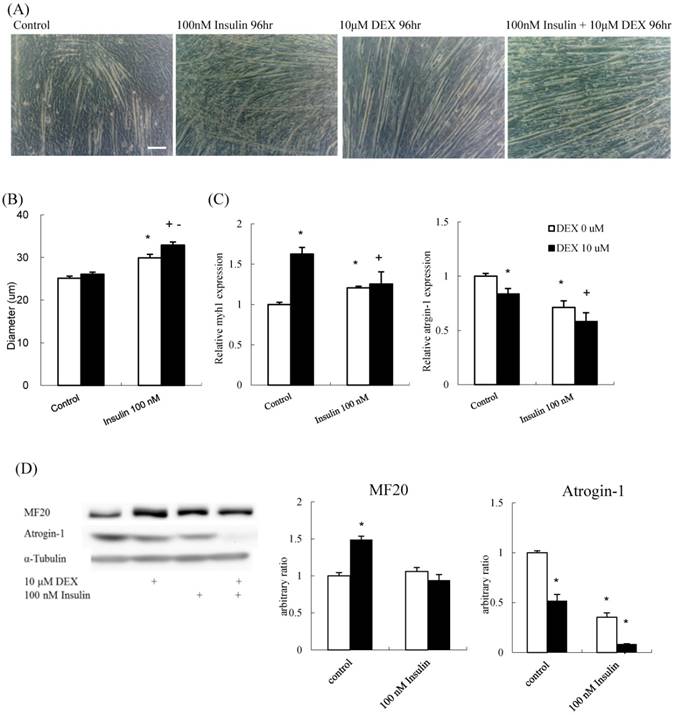
To determine if insulin and DEX have a synergistic effect on muscle differentiation, the diameter of myotubes treated with 100 nM insulin alone, 10 μM DEX alone, or a combination were determined. The myotube diameter increased in all the three experiment groups compared to that in the control (Figures 5A and B). The mRNA expression of myh1 increased, whereas that of atrogin-1 decreased, in all experimental groups compared to that in the control group (Figure 5C). The protein expression level of atrogin-1 decreased in all treatment groups. Myosin protein increased after treated with DEX; however, the effects of insulin and insulin plus DEX were not as expected (Figure 5D). Thus, DEX and insulin contributed to myoblast differentiation, individually or synergistically, and myotube diameter was thickest in the group treated with a combination of the two. Thus, new differentiation protocol combining DEX and insulin could augment the differentiation process.
The effect of DEX and insulin treatment on myoblast differentiation (A) Phenotypic change in myotubes. Scale bar: 100 μm. (B) The diameter of myotubes treated with 100 nM insulin, and 100 nM insulin combined with 10 μM DEX was increased compared to that of the control. (C) myh1 mRNA expression increased and atrogin-1 mRNA expression decreased in all treatment groups compared to that in the control group. (D) The protein expression level of atrogin-1 decreased in all treatment groups, whereas myosin heavy chain (MF20) increased only in the 10 μM DEX group. Results are presented as the mean ± SEM from three independent experiments. Asterisk (*) indicates significant difference compared to control. Plus sign (+) indicates significant difference compared to DEX only treatment. Minus sign (-) indicates significant difference between DEX and no DEX treatment groups. White bars indicate vehicle treatment; black bars indicate 10 μM DEX-combined treatment.
Discussion
To our knowledge, this is the first study to demonstrate that DEX not only causes muscle atrophy but also induces muscle hypertrophy, depending on the differentiation stage at which it was applied. That is, the effect of DEX is stage-dependent. DEX treatment at the myoblast stage induced muscle hypertrophy and enhanced myotube differentiation. On the other hand, DEX treatment at the myotube stage induced muscle atrophy.
Similar to the previous reports, we found that DEX treatment at the myotube stage has a dose-dependent atrophic effect on myotubes [9-11]. The increased expression of atrophic markers myostatin and atrogin-1, and decreased expression of the myoblast marker pax7, indicated that the atrophic process was caused by activation of the ubiquitin protease pathway, not by dedifferentiation.
In contrast, when DEX was added at the myoblast stage as pretreatment, the differentiation process was enhanced. We observed thicker myotube diameters, increased myh1 expression, and decreased myostatin and atrogin-1 expression. The proteolytic pathway was inhibited, and the terminal differentiation protein, myosin heavy chain, was increased 1.7-fold. We showed that DEX pretreatment had an anabolic and pro-differentiation effect, which was seldom discussed previously. This finding could partly explain the fact that glucocorticoid-induced myopathy is less in the children [12], since more myoblasts exist in the young than in the adult muscle. There are few studies reporting a pro-differentiation effect of DEX on different cell lines. As early as 1980, Guerriero et al found that DEX increased myoblast proliferation and low extent of muscular differentiation at differentiating myoblast [13]. The main difference is that their myoblast received persistent DEX treatment, not pre-treatment for 3 days only as in our study. Grigoriadis et al reported that DEX enhanced differentiation of bone-derived clonal cells toward a muscular lineage in a dose-dependent manner [14]. They also reported a differential effect based on different treatment durations, but not based on treatment at different stages. Using creatine kinase (CK) activity as a terminal differentiation marker, another study by Whitson et al demonstrated that DEX increased CK activity in C2C12 myoblasts in a dose-dependent manner. They proposed that glucocorticoid effects on CK activity were modulated by the maturity of the cells [15].
Despite its systemic side effect, corticosteroid is frequently locally injected to treat muscle injury. Hakim et al. reported that single injection of dexamethasone was beneficial to muscle strain by decreasing local inflammatory cytokines and increasing contractile tension. No significant histological muscle atrophy was observed after one dose dexamethasone administration [16]. Upon injury, the satellite cells, dormant myoblasts resided by sarcolemma, will become active and fuse to form muscle fiber. We think that corticosteroid has dual effects to keep muscle in balance state. One is to enhance myoblast differentiation and the other is to cause myotube atrophy. Thus, local and short-term administration of corticosteroids might be beneficial to acute muscle injury. Future studies are needed to find the optimal timing and dose in vivo for muscle strain treatment.
C2C12 is an immortal line of mouse skeletal myoblasts originally derived from satellite cells of the thigh muscle [17]. A general principle for myoblast differentiation is contact inhibition and serum deprivation [18], followed by division, elongation, and fusion. Many studies have treated differentiated myotubes with DEX and showed atrophic effects [9, 11, 19]. The molecular pathways involved in the atrophic process include the ubiquitin-proteasome system, the lysosomal system, and the calcium-dependent system [20]. As for the pro-differentiation effect of DEX, the mechanism remains unclear. Although DEX has also been used for decades to differentiate MSCs into adipogenic, chondrogenic, and osteogenic lineages [21], it has not been used for muscular differentiation [22].
Myostatin is a negative regulator of muscle growth and converts slow oxidative (type I) fibers to fast glycolytic (type IIB) fibers. It is expressed mainly in the type II fibers [23]. Muscle-specific myostatin transgenic mice showed increased expression of corticosteroids and moderate muscle atrophy [24]. The molecular mechanism involved was shown to be the induction of the ubiquitin-proteasome system, which is also the molecular pathway controlling DEX-induced muscle atrophy [25]. Fluorinated steroids including DEX induced selective type IIb (fast-twitch glycolytic) fiber atrophy [20]. This could explain why the increase in myostatin expression was lower in the high concentration DEX treatment group compared to that in the low concentration group (Figure 1C). In addition, myostatin-producing type II fibers were more diminished in the high concentration group.
Insulin induces myogenesis in C2C12 through the PI 3-kinase/p70S6-kinase and p38-MAPK pathways [26]. Previously, some myocyte differentiation protocols have contained 1 μM insulin [18, 27], whereas others did not [9, 11]. When we added insulin and DEX simultaneously, the myotube diameter increased more dramatically than with either treatment alone. The content of myosin heavy chains also increased. It is reasonable that insulin could enhance myogenic differentiation because it is a very important anabolic hormone that increases protein synthesis and cell growth, and is a common pro-differentiation factor. Thus, we propose a new formula, containing both 10 μM DEX and 100 nM insulin, for myoblast differentiation. We hope that this new differentiation protocol will facilitate faster and better differentiation in related experiments in the future.
This study had some limitations. We measured the protein content of myosin heavy chains because it is most abundant in myocytes. However, the expression of other differentiation markers, both early and terminal, such as MyoD, myogenin, creatine kinase or cytoskeleton proteins was not tested. Second, it remains to be determined if the hypertrophic effect is universal to all myogenic cells from different species, such as L6 myoblasts from rat [9] and human skeletal myoblast [28]. Experiments employing other myogenic cell lines could answer this question. Furthermore, the molecular mechanism controlling DEX-induced myocyte hypertrophy demands future investigation.
We demonstrated that DEX treatment at the myoblast stage could facilitate myoblast differentiation and significantly increase myotube diameter. A new protocol for C2C12 myoblast differentiation utilizing both DEX and insulin was proposed in this study.
Acknowledgements
This study was sponsored by a grant from the National Science Council (NSC 102-2314-B-002 -013).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Askari A, Vignos Jr PJ, Moskowitz RW. Steroid myopathy in connective tissue disease. Am J Med. 1976;61:485-92
2. Inder WJ, Jang C, Obeyesekere VR, Alford FP. Dexamethasone administration inhibits skeletal muscle expression of the androgen receptor and IGF-1 - implications for steroid-induced myopathy. Clin Endocrinol. 2010;73:126-32
3. Schakman O, Gilson H, Thissen JP. Mechanisms of glucocorticoid-induced myopathy. J Endocrinol. 2008;197:1-10
4. Pereira RMR, Freire de Carvalho J. Glucocorticoid-induced myopathy. Joint Bone Spine. 2011;78:41-4
5. Das M, Wilson K, Molnar P, Hickman JJ. Differentiation of skeletal muscle and integration of myotubes with silicon microstructures using serum-free medium and a synthetic silane substrate. Nat Protocols. 2007;2:1795-801
6. Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258-70
7. Han DS, Huang HP, Wang TG, Hung MY, Ke JY, Chang KT. et al. Transcription activation of myostatin by trichostatin A in differentiated C2C12 myocytes via ASK1-MKK3/4/6-JNK and p38 mitogen-activated protein kinase pathways. J Cell Biochem. 2010;111:564-73
8. Wang M, Amano SU, Flach RJR, Chawla A, Aouadi M, Czech MP. Identification of Map4k4 as a novel suppressor of skeletal muscle differentiation. Mol Cell Biol. 2013;33:678-87
9. Menconi M, Gonnella P, Petkova V, Hasselgren S-O. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J Cell Biochem. 2008;105:353-64
10. Marinovic AC, Zheng B, Mitch WE, Price SR. Ubiquitin (UbC) expression in muscle cells is increased by glucocorticoids through a mechanism involving Sp1 and MEK1. J Biol Chem. 2002;277:16673-81
11. Desler MM, Jones SJ, Smith CW, Woods TL. Effects of dexamethasone and anabolic agents on proliferation and protein synthesis and degradation in C2C12 myogenic cells. J Anim Sci. 1996;74:1265-73
12. Aulakh R, Singh S. Strategies for minimizing corticosteroid toxicity: a review. Indian J Pediatr. 2008;75:1067-73
13. Guerriero VJ, Florini JR. Dexamethasone Effects on Myoblast Proliferation and Differentiation. Endocrinology. 1980;106:1198-202
14. Grigoriadis AE, Heersche JN, Aubin JE. Differentiation of muscle, fat, cartilage, and bone from progenitor cells present in a bone-derived clonal cell population: effect of dexamethasone. J Cell Biol. 1988;106:2139-51
15. Whitson PA, Stuart CA, Huls MH, Sams CF, Cintron NM. Dexamethasone effects on creatine kinase activity and insulin-like growth factor receptors in cultured muscle cells. J Cell Physiol. 1989;140:8-17
16. Hakim M, Hage W, Lovering R, Moorman III C, Curl L, DeDeyne P. Dexamethasone and recovery of contractile tension after a muscle injury. Clin Orthop Relat Res. 2005:235-42
17. Burattini S, Ferri P, Battistelli M, Curci R, Luchetti F, Falcieri E. C2C12 murine myoblasts as a model of skeletal muscle development: morpho-functional characterization. Eur J Histochem. 2004;48:223-34
18. Blau HM, Chiu CP, Webster C. Cytoplasmic activation of human nuclear genes in stable heterocaryons. Cell. 1983;32:1171-80
19. Son YH, Lee SJ, Lee KB, Lee JH, Jeong EM, Chung SG. et al. Dexamethasone downregulates caveolin-1 causing muscle atrophy via inhibited insulin signaling. J Endocrinol. 2015;225:27-37
20. Gupta A, Gupta Y. Glucocorticoid-induced myopathy: Pathophysiology, diagnosis, and treatment. Indian J Endocrinol Metab. 2013;17:913-6
21. Oshina H, Sotome S, Yoshii T, Torigoe I, Sugata Y, Maehara H. et al. Effects of continuous dexamethasone treatment on differentiation capabilities of bone marrow-derived mesenchymal cells. Bone. 2007;41:575-83
22. Yuasa M, Yamada T, Taniyama T, Masaoka T, Xuetao W, Yoshii T. et al. Dexamethasone enhances osteogenic differentiation of bone marrow- and muscle-derived stromal cells and augments ectopic bone formation induced by Bone Morphogenetic Protein-2. PLoS ONE. 2015;10:e0116462
23. Hennebry A, Berry C, Siriett V, O'Callaghan P, Chau L, Watson T. et al. Myostatin regulates fiber-type composition of skeletal muscle by regulating MEF2 and MyoD gene expression. Am J Physiol Cell Physiol. 2009;296:C525-C34
24. Reisz-Porszasz S, Bhasin S, Artaza JN, Shen R, Sinha-Hikim I, Hogue A. et al. Lower skeletal muscle mass in male transgenic mice with muscle-specific overexpression of myostatin. Am J Physiol Endocrinol Metab. 2003;285:E876-E88
25. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25-39
26. Conejo R, Valverde AM, Benito M, Lorenzo M. Insulin produces myogenesis in C2C12 myoblasts by induction of NF-κB and downregulation of AP-1 activities. J Cell Physiol. 2001;186:82-94
27. Casas-Delucchi CS, Brero A, Rahn H-P, Solovei I, Wutz A, Cremer T. et al. Histone acetylation controls the inactive X chromosome replication dynamics. Nat Commun. 2011;2:222
28. Cheng CS, El-Abd Y, Bui K, Hyun Y-E, Hughes RH, Kraus WE. et al. Conditions that promote primary human skeletal myoblast culture and muscle differentiation in vitro. Am J Physiol Cell Physiol. 2014;306:C385-C95
Author contact
![]() Corresponding author: Dr. Der-Sheng Han, Department of Physical Medicine and Rehabilitation, National Taiwan University Hospital BeiHu Branch. No. 87, Nei-Jiang Street, Taipei 108, Taiwan. Tel: 886-2-23717101 ext. 5001 Fax: 886-2-23758363 E-mail: dshan1121com.tw
Corresponding author: Dr. Der-Sheng Han, Department of Physical Medicine and Rehabilitation, National Taiwan University Hospital BeiHu Branch. No. 87, Nei-Jiang Street, Taipei 108, Taiwan. Tel: 886-2-23717101 ext. 5001 Fax: 886-2-23758363 E-mail: dshan1121com.tw