Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Biological characteristics of...
CTC detection platforms for...
Pancreatic CTCs in clinical...
Pancreatic CTCs in basic research
ctDNA in clinical application
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2016; 13(12):902-913. doi:10.7150/ijms.16734 This issue Cite
Review
Circulating Tumor Cells and Circulating Tumor DNA Provide New Insights into Pancreatic Cancer
Yang Gao, Yayun Zhu, Zhou Yuan ![]()
Department of General Surgery, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, People's Republic of China
Received 2016-7-5; Accepted 2016-9-13; Published 2016-11-4
Abstract
Pancreatic cancer has a rather dismal prognosis mainly due to high malignance of tumor biology. Up to now, the relevant researches on pancreatic cancer lag behind seriously partly due to the obstacles for tissue biopsy, which handicaps the understanding of molecular and genetic features of pancreatic cancer. In the last two decades, liquid biopsy, including circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA), is promising to provide new insights into the biological and clinical characteristics of malignant tumors. Both CTCs and ctDNA provide an opportunity for studying tumor heterogeneity, drug resistance, and metastatic mechanism for pancreatic cancer. Furthermore, they can also play important roles in detecting early-stage tumors, providing prognostic information, monitoring tumor progression and guiding treatment regimens. In this review, we will introduce the latest findings on biological features and clinical applications of both CTCs and ctDNA in pancreatic cancer. In a word, CTCs and ctDNA are promising to promote precision medicine in pancreatic cancer.
Keywords: Circulating tumor cells, Circulating tumor DNA, Pancreatic cancer, Precision medicine, Metastasis, Drug resistance, Tumor heterogeneity
Introduction
Pancreatic cancer is one of the most devastating malignant tumors with a 5-year survival rate of approximately 5% and increasing incidence rate, which is the seventh leading cause of cancer related death in both men and women worldwide [1-3]. In 2015, about 48,960 new cases are expected to occur and about 40,560 people are expected to die from pancreatic cancer in USA [4]. In China, the incidence of pancreatic cancer has reached 14-17 per 100,000 people in some area [5]. What's worse, the annual mortality of pancreatic cancer almost equals to the morbidity. The poor prognosis of pancreatic cancer is mainly associated with delayed diagnosis, deep anatomic location and non-specific symptoms. At present, surgical resection is the only potentially curative treatment for pancreatic cancer. Unfortunately, only 15%-20% of patients are candidates for pancreatectomy at the time of diagnosis [2]. Tissue biopsy is the golden standard for the diagnosis of pancreatic cancer for those patients without surgery or before neoadjuvant therapy administration. However, there are many obstacles for tissue biopsy, including potential surgical complications, tumor dissemination, and false negative results [6, 7]. In addition, sufficient material from primary tumors in pancreatic cancer is scarce as the majority of patients present with advanced disease and only biopsy material is available and thus CTC and ctDNA can help fill this gap in order to perform the genomic analysis.
Recently, liquid biopsy, as a less invasive approach, is becoming the research hotspot and attracts much attention owing to remarkable advantages. The broad conception of liquid biopsy includes circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), circulating microRNAs, circulating proteins, extracellular vesicles and so on [8, 9]. Particularly, CTCs and ctDNA are crucial components in the realm of liquid biopsy. CTCs and ctDNA have several prominent characteristics for cancer managements: (1) assessing risk factor and achieving early diagnosis; (2) monitoring treatment response and drug resistance dynamically; (3) providing prognostic information by evaluating relapse and metastatic risk; (4) opening a window for studying tumor heterogeneity and evolution procedure; (5) helping to understand the tumorigenesis, recurrence and metastasis [10, 11]. Recently, a great deal of attention has been focused on pancreatic cancer and some remarkable findings on biological underpinnings have been made via CTCs and ctDNA [9, 12, 13]. In this review, we will summary the relevant studies on CTCs and ctDNA and their potential applications in managements of pancreatic cancer.
Biological characteristics of CTCs and ctDNA
CTCs are shedding from both primary and secondary tumors into bloodstream [14]. CTCs are generally more likely to be detected in advanced tumors due to higher tumor burden. Meanwhile, CTCs could also appear unexpectedly early in the disease process, after primary radical treatment, and even when clinically detectable tumor or recurrence doesn't appear [15, 16]. Because the half-time of CTCs is quite short (1-2.4 hours), they could reflect the current status of both primary tumors and secondary deposits accurately and sensitively [17]. In particular, a subset of CTCs have the phenotypes of cancer stem cells (CSCs), which may initiate tumor formation and drug resistance [18]. The mutual transformation of CTCs and CSCs are linked by epithelial-to-mesenchymal transition (EMT) process (Figure 1) [19]. CTCs bear great potential for early diagnosis, treatment monitoring, and predicting prognosis for various cancer types. With the development of detection technology of CTCs, some sophisticated and exquisite devices have been developed for efficient enrichment and identification of CTCs, especially for viable CTCs, which paves the way for further exploration of tumor heterogeneity, tumor metastasis, and drug resistance [20, 21].
Release of CTCs and ctDNA into the circulation.
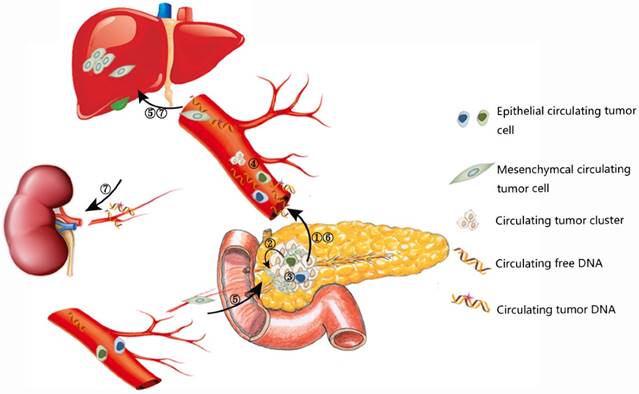
Similar to CTCs, ctDNA provides another approach for monitoring tumor genome as a less invasive approach and it has unique features compared to CTCs. ctDNA is generally considered to be released from necrotic, apoptotic cells in primary tumors, secondary deposits and CTCs [9, 22, 23]. As a fraction of cell free DNA (cfDNA) with genetic mutations (range from 0.01 % and more than 90%, usually 1.0%), ctDNA represents an average of DNA released by all tumor cells, so it has the potential to reflect the entire tumor burden [24, 25]. For pancreatic cancer, global genomic sequencing of 24 patients revealed an average of 63 genic alterations associated which defined 12 core cellular signaling pathways [26] and the average number of mutated genes in pancreatic cancer ranged from 26 to 42 [27, 28]. In regard to specific mutation type, a recent proof-of-concept study including 99 patients demonstrated that approximately 90% genetic variations were point mutations and small indels, the rest were mainly structural variants [29]. Consistent with the previous studies, the most prevalent mutated genes proved to be KRAS, TP53, SMAD4, CDKN2A and ARID1A. Besides, novel candidate drivers of pancreatic carcinogenesis (KDM6A and PREX2) were also identified [26, 28]. A series of genetic variations lead to carcinogenesis and development of pancreatic cancer. When the mutated DNA was released to bloodstream in a passive or active way, they could be detected by different methods and the detected mutated DNA could well reflect the tumor genomic landscape [25, 30], so the underling genetic changes revealed by novel sequencing technology will accelerate the development of liquid biopsy.
CTC detection platforms for pancreatic cancer
Many clinical and preclinical studies on pancreatic CTC have been performed via various devices (summarized in Table 1). Notably, the classic EpCAM-dependent CellSearch system rendered limited detection rate for pancreatic cancer (11% in localized advanced pancreatic cancer and 19% in metastatic pancreatic cancer) [31, 32]. The relative low CTC number may result from three reasons. (1) CTCs get trapped in liver as blood flows though portal vein into systematic circulation [33]. (2) The blood flow decreases by 60% in malignant pancreatic tumors compared with normal pancreatic tissues, so fewer tumor cells had the chance to invade into the bloodstream [34]. (3) The process of EMT decreased expression of epithelial markers, such as E-cadherin and EpCAM, making them undetectable by epithelial marker-dependent approaches [15, 35]. Several modified device have been developed for better detection of pancreatic CTCs. Immuno-FISH platform is a negative-enrichment method for CTC detection and our preliminary results showed that the sensitivity could reach 100% by combining CTC and CA19-9 [36]. The PCR-based strategy have also been reported to detect pancreatic CTCs, but the platform may produce false-positive results [37, 38]. The size-based filtration devices could potentially overcome some limitations in other platforms and has achieved satisfactory results in pancreatic cancer [39, 40]. This approach provides an exciting potential strategy for studying the mechanism of metastases, and predicting clinical outcome by separating both epithelial and mesenchymal CTCs, culturing viable and virgin CTCs [40, 41]. Furthermore, CTC captured by sized-based platform can be validated by looking for tumor specific mutations such as a KRAS mutation which occurs in up to 95% of primary tumors [42, 43].
Summary of clinical studies on CTCs in pancreatic cancer
| Reference | Positive criteria | Positive rate | Mean±SD | No. of patients | Median OS with vs without | Technique |
|---|---|---|---|---|---|---|
| Z'Graggen, et al, 2001 [57]. | AE1/AE3-positive | 26% | NR | 27/105 | NS (P=0.35) | Immunocytochemical assay |
| Allard, et al, 2004 [31]. | ≥2 CTCs/7.5 ml | 19% | 2±6/7.5 ml | 4/21 | NR | CellSearch system |
| Nagrath, et al, 2007 [45]. | ≥5 CTCs/ ml | 100% | 196±228/ ml | 15/15 | NR | CTC chip |
| Kurihara, et al, 2008 [58]. | ≥1 CTC/7.5 ml | 57% | 22.8±35.0/7.5 ml | 8/14 | 52.5 vs 308.3 days (P<0.01) | CellSearch system |
| Zhou, et al, 2011 [59]. | EpCAM-positive | 100% | NR | 25/25 | NR | RT-PCR |
| Khoja, et al, 2012 [60]. | ≥1 CTC/7.5 ml ≥1 CTC/7.5 ml | 39% 89% | 6/7.5 ml 26/7.5 ml | 21/54 24/27 | 164 vs 127 days (P=0.19) NS (P=0.36) | CellSearch system ISET |
| Bidard, et al, 2013 [32]. | ≥1 CTC/7.5 ml | 11% 50% | 2.7±4.6/7.5 ml 8 and 44/7.5 ml | 11/79 2/4 | 11 vs 13 months (P=0.01) NR | CellSearch system |
| Sheng, et al, 2014 [61]. | ≥1 CTC/ml | 94% | 2.8±1.8/ml | 17/18 | NR | GEM chip |
| Bobek , et al, 2014 [41]. | Cytomorphological features | 66.7% | NR | 16/24 | NR | MetaCell technology |
| Cauley, et al, 2015 [62]. | Positive-stained | 49% | NR | 51/105 | NS (P=0.69) | ScreenCell device |
| Zhang, et al, 2015 [63]. | ≥2 CTCs/ 3.5ml | 68.18% | 7.4±13.9/3.5 ml | 15/22 | NR (P=0.0458) | Immuno-FISH |
| Kulemann, et al, 2016 [39]. | Positive-stained and KRAS mutation | 86% | NR | 18/21 | 16 vs 10 months (P=NS) | ScreenCell device |
| Katherine, et al, 2016 [40] | ≥1 CTC/ml | 78% | 30/ml | 39/50 | 13.7 vs not reached (P=0.008) | ISET |
Abbreviations: SD, standard deviation; OS, overall survival; NR, no reports of CTC number or overall survival; NS, no significant difference, ISET, isolation by size of epithelial tumor cells; GEM chip: geometrically enhanced mixing chip.
Microfluidic devices, including CTC-chip, HB-chip, CTC-iChip, have shown great promise for CTC enumeration and function analysis [44-46]. In the preclinical studies, the detection rate of various cancer types by the microfluidic devices could reach as high as 90% [44-46], but the detection rates of CTCs in later researches were lower in both localized and metastatic tumors than previous reported results [47]. Despite the unsatisfactory fact, about 98% of CTCs captured by these microfluidic devices maintained viable since the blood specimen needn't excessive pretreatments [45]. Therefore, these viable and intact CTCs could be utilized for genome analysis, expression analysis, protein analysis and functional analysis [48]. In particular, the relevant researches are remarkable in tumor dissemination [13, 15, 27], drug resistance [49, 50], and function analysis in CTC-derived explants [51] on several cancer types, including pancreatic cancer [13, 15].
Pancreatic CTCs in clinical research
Early diagnosis of pancreatic cancer
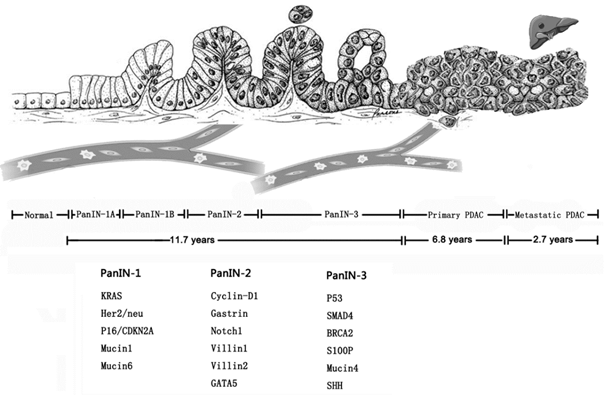
Most pancreatic cancer can't be radically removed because of delayed diagnosis, thus, it is crucial to find specific and efficient biological marker of pancreatic cancer for early diagnosis. Pancreatic cancer is driven by a subgroup of underlying genetic mutations, including KRAS, CDKN2A, SMAD4 and TP53 [26, 52]. By analyzing genetic evolution of pancreatic cancer, one model showed that the total disease course of pancreatic cancer was almost 20 years, and if so, there would be enough time to carry out intervening measures to improve the clinical outcomes (Figure 2) [53]. At the early stage of tumor formation, even before tumor formation, CTCs could be detected in the peripheral blood. PanIN, intraductal papillary mucinous neoplasms (IPMN) and mucinous cystic neoplasm (MCN) can be evolved into pancreatic cancer, therefore, they are usually regarded as premalignant lesions [2, 54]. In the pancreatic intraepithelial neoplasia (PanIN) mouse models, although micro- or macrometastasis didn't occur, liver seeding could be detected and the single cells were located in the blood vessels with distinguished marker [15]. In another study, Andrew Rhim et al. confirmed that cancer cells could enter circulation before tumor detection by studying patients with IPMN and MCN [55]. The early dissection of pancreatic cancer necessitates sensitive and accurate technique to detect and determine the tumor biology of pancreatic cancer. Although it has been reported CTCs could realize early diagnosis for lung cancer developed from chronic obstructive pulmonary disease (COPD), similar results in pancreatic cancer hasn't been reported [56]. Therefore, devices for CTC capture with high sensitivity and specificity and some prospective studies are required to validate the clinical significance of early detection of CTCs in pancreatic cancer.
Genetic changes in multistep progress model in pancreatic cancer.
Treatment monitoring
Since half-life of CTCs is quite short, CTCs could monitor the cancer progress dynamically in real-time [17]. The accurate knowledge of disease evolution will be of utmost importance for treatment decision. For pancreatic cancer, neoadjuvant therapy could not only downstage the primary tumor to improve the resectability, but also test the response to treatment regimen to avoid the delayed treatment or chemotherapy after surgery [76]. In general, the conventional imaging examinations usually lag behind the evolution of tumor biology. However, CTCs could provide the accurate scenario of tumor progress and the best time for surgery can be determined by noting the drop of CTCs [77]. In addition, the dynamical changes of CTC enumeration were closely associated with the radiographic tumor response and CTCs could well reflect the genetic information of primary tumor [78]. This phenomenon has been validated in non-small lung cancer and whether it could be applied to other cancer types remains to be confirmed [79]. However, the SWOG S0500 study demonstrated that treatment modification according to the CTC enumeration didn't produce prolonged overall survival or progress free survival [80]. This indicated that necessity of treatment modification and how to swift treatment regimen should be taken into consideration for better clinical efficiency.
Prognostic information
CTCs can be regarded as the seeds for distant dissemination of various cancers. It's reported that only about 2.5% of CTCs would result in micrometastasis and as few as 0.01% would finally develop into macrometastasis which lead to disease recurrence and mortality [81, 82]. Therefore, progress free survival would be direct indicator of the function of these CTCs [83]. Some studies found relationship between CTC enumeration and prognostic information of pancreatic cancer [31, 32, 63, 84], while some didn't [57, 60, 62]. A recent meta-analysis including 623 pancreatic cancer patients revealed that the patients with positive-CTC had worse progress free survival (HR=1.23, 95 %; CI=0.88-2.08, P<0.001) and overall survival (HR=1.89, 95 % CI=1.25-4.00, P<0.001) [85]. The discrepancies on the prognosis information rise an issue whether these isolated cells have malignant biological characteristics or whether they are just a tip of the iceberg [10]. For example, a small pilot study validated that EpCAM+ CTCs indicated poor outcome among cancer patients, whereas the EpCAM- CTCs was not associated with poor overall survival, so a series of analysis should be carried out to study the tumorigeneity of these isolated cells at various levels to provide in-depth explanations of prognostic information [86, 87].
Pancreatic CTCs in basic research
Interpreting tumor heterogeneity
Tumor heterogeneity is a main barrier to conquer cancer. The interpatient tumor heterogeneity has been well studied while intrapatient tumor heterogeneity needs more attention which is responsible for drug resistance [10, 88]. Mutational analysis and genomic rearrangements reveal genomic instability, the genetic evolution of pancreatic cancers, temporal and spatial heterogeneity in both primary and secondary pancreatic cancer [89]. Tumor heterogeneity can be generally divided into four classifications: intratumoral heterogeneity, intermetastatic heterogeneity, intrametastatic heterogeneity, and interpatient heterogeneity [90]. Since CTCs act as a bridge between primary tumor and secondary tumors and they are the seeds of metastasis and tumor self-seeding, the underling mechanism on tumorigenesis, metastasis, and drug resistance can be obtained by analyzing CTCs, thus avoiding the complexity of tumor heterogeneity, which will be conductive to finding new therapeutic targets and promote targeted therapy in return [91, 92].
Deciphering drug-resistance
CTCs also function as a powerful weapon to decipher the acquired drug resistance mechanisms and guide rational use of medicines. Miyamoto et al. reported the research findings of RNA-Seq of single prostate CTCs isolated by CTC-iChip [50]. The analysis of CTCs from patients undertaking androgen receptor inhibitor and untreated cases revealed the activation of noncanonical Wnt signaling, which was involved in multiple downstream regulators of cell survival, proliferation, motility and the maintenance of stem cell populations [93]. By deep analysis of CTCs, more pathogenic mechanism and potential therapeutic targets could be revealed. The application of next-generation sequencing (NGS) for CTCs will provide tumor information in real-time and guide following therapeutic regimen earlier, meeting the targeted therapy for cancer [94]. In another concept-of-proof study, breast CTCs were captured by CTC-iChip and then one or more cell lines were successfully generated from 6 patients of 36 patients [49]. Notably, the captured CTCs shared cytological characteristics with matched primary tumors and xenograft tumor in immunosuppressed non-obese diabetic scid gamma (NSG) female mice model and similar results were found in small cell lung cancer and colorectal cancer patients by CellSearch system [51, 95]. Therefore, genotyping and functional testing for drug susceptibility in CTCs could accurately reflect these features in primary tumors. For example, optimal treatments for breast cancer patients with ER-positive and ESR1 mutation is unknown, HSP90 inhibitor STA9090 alone could demonstrate cytotoxicity and a low dose of STA9090 indeed suppressed the ER level, which provided an opportunity to offer optimal therapies for cancer patients during the disease course [49].
Explaining the metastatic mechanism
CTCs could help to explain the metastatic mechanism of pancreatic cancer. Single-molecule RNA sequencing of captured pancreatic CTC using HbCTC-Chip method proved that noncanonical Wnt signaling pathway may contribute metastatic dissemination in human pancreatic cancer [13]. In this study, Wnt2 mRNA was frequently observed in CTCs and metastatic ascites cells while seldom expressed in the primary tumor tissue. Besides, Wnt2 in pancreatic cancer cells had the ability to suppress anoikis, enhance anchorage-independent sphere formation, and increase metastatic propensity in vivo. Then therapy targeted Wnt signaling, such as TGF-b activated kinase 1 (TAK1), could inhibit pancreatic cancer metastasis [96]. In another study, identified pancreatic CTCs using epitope-independent microfluidic capture were analyzed by single-cell RNA sequencing, and extracellular matrix genes, which were responsible for cell migration and invasiveness were found to be highly expressed [12]. This discovery cast light on the metastatic mechanism and the design the proper agents to prevent distant dissemination.
CTCs are found in the circulation as either single CTCs or CTC clusters. As a form of CTCs, CTC clusters were relatively infrequent compared to single CTCs (2.6% versus 97.6%), but the metastatic capability of CTC clusters was as much as 50 times of single CTCs in breast cancer [27]. Besides, CTCs also travel with other cells in circulation such as macrophages and neutrophils that help protect the CTCs whilst in the blood stream. This is one reason why they are not detected very efficiently with some of the CTC detection systems on the market [97]. The presence of CTC clusters predicated a poor clinical outcome in many cancers including lung, breast, or colorectal cancers [27, 98, 99]. CTC clusters were also observed in the blood stream of pancreatic cancer patients, the in-depth implications remained to be discovered [13]. Single-cell RNA sequencing revealed that expression of plakoglobin, which played a pivotal role in the regulation of cell-cell adhesion and Wnt signaling pathway increased about 200 times in CTC clusters compared with single CTCs [27, 100]. In conclusion, researches in molecular characterization of CTCs and CTC clusters yield novel and profound insights into metastatic mechanism and then targeted drugs can be designed to intervene in corresponding signaling paths.
ctDNA in clinical application
The clinical application of ctDNA was initially studied in 1998 using quite conventional mutant allele-specific amplification method and in recent five years more and more studies were carried out with sophisticated sequencing technology, such as digital PCR, next-generation sequencing [101]. The clinical applications of ctDNA in pancreatic cancer are quite intriguing and important, but the results are also quite mixed, which need further verification and reconsideration. The relevant studies were summarized in Table 2.
Summary of clinical studies on ctDNA in pancreatic cancer
| Reference | Origin | Technique | Number of PC patients | Results | Main findings |
|---|---|---|---|---|---|
| Terumasa Yamada et al., 1998 [64] | Plasma | Mutant allele-specific amplification | 21 I:2, II:2; III:2; IV:13 | 60% of patients with K-ras mutations in tissue showed identical mutations in plasma | ctDNA may be useful for evaluating tumor burden and treatment efficiency |
| Antoni Castells et al., 1999 [65] | Plasma | Single-Strand Conformation Polymorphism | 44 I:4, II:11; III:5; IV:23 | Mutant K-ras was found in 27% of plasma samples | Mutant-type KRAS was associated with shorter survival time |
| Feng Dianxu et al., 2002 [66] | Plasma/ tissue | Direct sequencing | 41 I:2, II:6; III:5; IV:26;NA:2 | ctDNA was detected in 70.7% of PCs | Plasma KRAS mutation analysis combined with serum CA19-9 determination could detect the majority of cases of pancreatic carcinoma |
| Uemura Takanori et al., 2004 [67] | Plasma/ tissue | Mismatch ligation assay | 28 I:2, II:8; III:7; IV:11 | ctDNA was detected in 35% of PCs | Genetic alterations present in the tumors of pancreatic cancer patients can be detected in their plasma |
| Rodolfo Marchese et al., 2006 [68] | Plasma/ tissue | Direct sequencing | 30 I:3, II:22; III:3; IV:2 | Media 333 ng/mL (125-525 ng/mL) | K-ras mutations were detected in 70% of neoplastic tissue samples, but no mutated DNA resulted in circulating DNA samples |
| Jan Da ¨britz et al.,2009 [69] | Plasma | Real-time PCR | 56 | KRAS mutations could be detected in 36% of PCs | The combination with CA 19-9 and KRAS mutation could improve the sensitivity or the diagnosis of PC |
| H. Chen et al., 2010 [70] | Plasma | Direct sequencing | 91 | KRAS mutation rate: 33% | KRAS mutation was correlated with clinical outcome in unresectable pancreatic cancers |
| Bettegowda C, et al., 2014 [22] | Plasma | Digital PCR | 155 | ctDNA was detectable in >75% of PC patients | ctDNA is an applicable biomarker that for a variety of clinical and research purposes |
| Oliver A.Zill, et al., 2015 [30] | Plasma /tissue | NGS | 18 | Diagnostic accuracy of cfDNA sequencing was 97.7% | cfDNA sequencing is feasible, accurate, and sensitive in identifying tumor-derived mutations and could guide targeted therapy. |
| Julie Earl et al., 2015 [71] | Plasma | ddPCR | 31 | KRAS mutation rate: 26% | KRAS mutant cfDNA was correlated strongly with overall survival |
| Erina Takai et al., 2015 [72] | Plasma | ddPCR and NGS | I-III:95 IV:163 | Media: I-III, 17.59 ng/2ml; IV, 21.65 ng/2ml | Potentially targetable somatic mutations were identified in 29.2% of patients examined by targeted deep sequencing of cfDNA. |
| Mark Sausen et al., 2015 [73] | Plasma | ddPCR | II :51 | Mutation rate: 43% | Detection of ctDNA after resection predicts clinical relapse and poor outcome, with recurrence by ctDNA detected 6.5 months earlier than with CT imaging |
| Hideaki Kinugasa et al., 2015 [74] | Serum | ddPCR | 131 | KRAS mutation rate: 54.5%-62.6% | KRAS mutations in ctDNA other than in tissue were associated with worse survival, especially in cases with a G12V mutation. |
| Tjensvoll K et al., 2016 [75] | Plasma | PNA-clamp PCR | locally advanced:2 metastatic:14 | KRAS mutation rate: 71% | The pre-therapy ctDNA was a predictor of both progression-free and overall survival. Changes in ctDNA levels corresponded both with radiological follow-up data and CA19-9 levels. |
Abbreviations: PC, pancreatic cancer; PNA-clamp PCR, peptide-nucleic acid clamp polymerase chain reaction; ctDNA, circulating tumor DNA; ddPCR, digital droplet PCR; NGS next-generation sequencing.
Early diagnosis of tumors
Analysis of SEER data suggests resectable pancreatic cancer has a dramatic survival advantage compared to unresectable pancreatic cancer (media survival: 36 months vs 7 months) [102], so early detection for higher resectability is very crucial for better clinical outcomes. Pancreatic cancer can be considered as an accumulative process of various genetic aberrations, and the mutated genes in the bloodstream will provide a clue of carcinogenesis of pancreatic cancer. Therefore, the less invasive and actionable ctDNA has great potential for pancreatic tumor screening among high-risk population (ie, a family history of pancreatic cancer, elder than 50 years, new-onset diabetes, smoking) [103, 104].
It has been reported that ctDNA could be detected in about 50% of early-stage pancreatic cancer by digital PCR approaches [22, 73]. Nevertheless, the whole exome sequencing identified an average of 26 mutations (range 1-116) in the tumor tissue in the early pancreatic cancer, so mutations could also be detected in the circulation theoretically because the genetic aberrations will be released in bloodstream [28]. Therefore, if more genetic mutations could be detected, the positivity of ctDNA may increase. To solve this issue, a conceptual “ctDNA-Chip” could be fabricated to assay more genes at a time and the mathematical modeling could be applied to evaluate the risk factor. When ctDNA is used as a diagnostic tool, several problems should be taken into consideration. Firstly, false-positive is a common problem of genetic diagnosis because many mutations appeared in both malignant and benign lesions and it's difficult to distinguish them solely by a single mutation [105, 106]. Secondly, the origin of ctDNA is difficult to determine because many mutations are shared by different tumors, such as KRAS, TP53 [107, 108]. In order to solve these problems, pancreatic cancer-specific gene markers should be discovered and the potential relationship of different genetic mutation should be revealed. Thirdly, these types of biomarkers, either CTC or ctDNA should be used in conjunction with imaging, as alone they are not 100% reliable. The problem of the overlap of genetic mutations in different cancer types is difficult to overcome. This said, the detection of a tumor associated mutations in KRAS or TP53, for example, in cfDNA may prompt a clinician to perform an imaging scan with the ability to detect a cancer in different anatomical locations not just in the pancreas which is also of clinical benefit.
Treatment Monitoring
Genetic variations in ctDNA could reflect the tumor tissues with considerable accuracy and feasibility [109, 110]. Since the genetic variations have many forms, ctDNA could be used to track tumor progress with higher specificity [111]. Besides, the half-time of ctDNA is only estimated to be about 2 hours in the body, so ctDNA could act as a flexible method to monitor the tumor progress dynamically [112]. Frank Diehl et al. have demonstrated that ctDNA in advanced colorectal cancer patients who received complete resection of all evident tumor tissue experienced a 99.0% of median decrease two to ten days after surgery [112]. In contrast, the patients undertaking incomplete resection showed slighter decreased or even increased level of ctDNA. Interestingly, the undetectable level of ctDNA 10 days after surgery in 4 patients predicted no recurrence, so similar to negative margin, “negative ctDNA” is also a key indicator for long-term survival [112]. Similar conclusions were drawn from relevant researches on early breast cancer patients undertaking curative resection. It was estimated that the detectable ctDNA at a single postsurgical time point predicated metastatic relapse with a hazard ratio of 25.1 [113]. What's more, ctDNA predicted the subsequent relapse with more accuracy compared to primary tumor, which was associated with genetic diversity in the development of micrometastatic disease before relapse [113]. For pancreatic cancer, detection of ctDNA after resection was a poor indicator for clinical relapse, and ctDNA detected clinical recurrence 6.5 months earlier than CT imaging [73]. As ctDNA and to a certain extent CTCs are markers of disease dissemination and relapse, they are important novel biomarkers for dynamic monitoring for these patients [114].
Providing prognostic information
The prognosis of pancreatic cancer is mainly determined by clinical presentations, tumor stage, histological characteristics, and the prognostic significance of ctDNA in pancreatic cancer is quite controversial [6, 115]. Generally speaking, the clinical utility of ctDNA for prognosis assessing is limited in the resected cases because the alteration of subsequent treatment options are variable and the genetic evolvement is random and unpredictable [53, 116]. Nevertheless, some potential genetic aberrations appearing in early-stage pancreatic cancers have been found to be associated with survival, which may play a decisive role in the disease progress and more work are warranted [28, 73]. In late-stage pancreatic cancer patients, ctDNA would also be of great help and it would provide thorough information for better management. Several pilot clinical researches have explored the potential prognostic function of ctDNA and the research objects of ctDNA were mainly popular point mutations [65, 112, 117].
Since KRAS gene mutation presents in about 90%-95% of pancreatic cancer and it is considered to be an early event in the tumorigenesis process since more than 90% of PanINs harbor KRAS mutations [118, 119]. 98% of KRAS mutations in PDAC occurs at position G12 and predominant substitution found at this position is G12D (51%), followed by G12V (30%) and G12R (12%) [118]. Therefore, KRAS attracts much attention and it proves to be a significant prognostic factor for survival [71, 120]. It has been demonstrated that ctDNA could be detected in about 50% and 90% of early-stage and late-stage pancreatic cancer patients, respectively, which is an essential condition as an excellent prognostic marker [22]. A pilot study enrolling 45 pancreatic ductal carcinoma patients at different disease stages showed KRAS mutation in the plasma correlated with a significantly worse overall survival (60 days for KRAS mutation positive vs 772 days for KRAS mutation negative) [71]. In this study, KRAS mutation was detected in 26% (8/31) of patients of all stages by droplet digital PCR and the majority mutation position was G12D (6/8), however, the patients with KRAS mutation was still relatively small and the specific determinant point mutation was difficult to identify. Hideaki Kinugasa et al. reported a higher sensitivity of KRAS mutation in serum (62.6%) by droplet digital PCR and they also find KRAS mutation in serum rather than tissue predicted a worse prognosis in both development set and validation set [120]. Another large-scale research also didn't find the prognostic significance of various G12 mutations in the tumor tissues [121]. These results may reflect the actual pathological progress in which the potent cells with aggressiveness and proliferation release more nucleic acid and this characteristics will help to identify the wrecker nucleic acid, thus resolving the tumor heterogeneity to some extent [120]. Furthermore, G12V mutation in serum was found to be correlated to a significantly shorter survival compared with G12D/G12R/wild-type [120]. The results were concordant with previous basic research findings that G12V mutation contributed to high invasive potential oncogenic activity [122, 123]. With the development of next-generation sequencing, more and more relevant genetic aberrations have been identified by clinical researches such as aschromatin-regulating genes MLL, MLL2, MLL3, ARID1A [73], SLIT receptor ROBO2, amplification at SEMA3A and PLXNA1 [28]. These clinical findings based on ctDNA help reveal valuable targets and then substantial pathologic mechanisms can be revealed by further researches of these targets of most interest, which finally provides proof-of-concept evidence for novel agents.
Managements of chemotherapy and targeted therapy
At present, chemotherapy drugs are usually uniformly administered despite the chemotherapeutic sensitivity. However, some patients will never relapse even without chemotherapy and some patients will relapse soon even with a certain chemotherapy regimen [124]. This situation calls for an accurate evaluation tool that could predict the individualized treatment response, thus avoiding overtreatment or futile treatment. With the advent of targeted therapy, the clinical indications and effect evaluation have been key issues for rational application of targeted drugs. ctDNA exhibits excellent characteristics to resolve the above issues in the era of targeted therapy. On the one hand, ctDNA could clarify the molecular marker of tumor tissue with satisfactory sensitivity and specificity, which could help to select optimal treatment [30, 110]. On the other hand, low level of ctDNA indicated a favorable prognosis [112, 125]. Therefore, it's feasible and practicable to administer treatment regimen according to genetic status by ctDNA. Several studies have demonstrated the potential of ctDNA in the cancer management [126]. In a recent clinical study, Oliver A. Zill et al. has demonstrated an intriguing case that EGFR deletion was detected in ctDNA 7 months earlier than tissue biopsy and the subsequent capecitabine and erlotinib lead to radiographic response and CA 19-9 normalization [30]. This phenomenon indicated that ctDNA could be used to guide targeted therapy, thus avoiding overtreatment and realizing precision medicine. Another example is BRAF mutation, which appears in about 2.2% of pancreatic tumors, and the targeted agent vemurafenib has been approved for metastatic melanoma with V600G aminoacid substitution in BRAF gene, so this subset of patients harboring such mutation may be susceptible to vemurafenib, which could be an alternative method for this lethal disease [127]. Nevertheless, several issues should be solved due to multiple genetic mutations and crosstalk of signaling pathways: development of multiple targeted drugs, identification of prognostic gene mutations, and selection of patients who will gain utmost benefit from specific targeted agents [128].
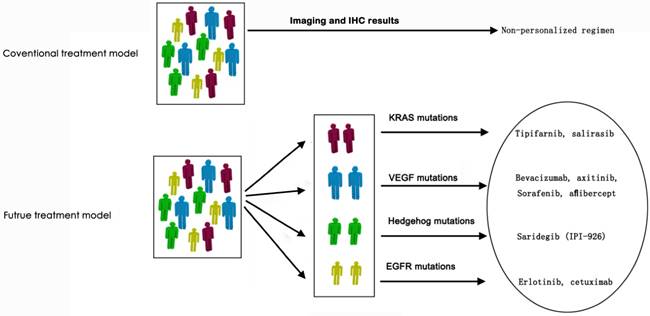
Targeted therapy has become standard therapy regimen for some tumors in the past 20 years, such as breast cancer, colorectal cancer, lung cancer, melanoma and so on [129, 130]. For pancreatic cancer, only erlotinib, an epidermal growth factor receptor inhibitor, is approved by FDA for clinical use [131]. However, the overall survival of gemcitabine plus erlotinib is 0.33 month longer than gemcitabine alone (median 6.24 months vs 5.91 months), so erlotinib hasn't been widely accepted in the management of pancreatic cancer due to the modest survival benefit and cost-effect margin [132, 133]. A potential reason for the unsatisfactory efficacy of targeted therapy in pancreatic cancer was lack identification of genomic profiling due to the inadequate biopsy for molecular characterization [30]. It has been reported that a KRAS-wild type in formalin-fixed and parrffin-embedded (FFPE) tumor samples correlated to a better overall survival (OS) under treatment regimen including erlotinib (median OS, 7.9 months in KRAS wild-type group and 5.7 months in KRAS mutation group; HR=1.68, P=0.005) [131]. The identified genetic status would be conductive to improving clinical outcomes. Besides, since more and more targeting signaling pathways in the epithelial compartments, targets in the stromal compartments and other potential targets have been discovered, the correspondent targeted agents, such as tipifarnib and salirasib targeted KRAS mutations, bevacizumab and sorafenib targeted to VEGF mutations, erlotinib and cetuximab targeted to EGFR mutations have been developed and some agents have resulted in decreased growth of pancreatic tumor in preclinical studies (Figure 3) [134-136]. Therefore, it's never more crucial to identify the genomic information of the individual pancreatic cancer.
Envisaged revolution of treatment model for pancreatic cancer in the era of precision medicine.
Conclusion
Pancreatic cancer is still a devastating disease, so the tumor biological features and clinical managements of pancreatic cancer require further intensive researches. CTCs and ctDNA are essential components of liquid biopsy and are promising to discover the hidden secrets of pancreatic cancer. Since CTCs and ctDNA are two independent entities, they are complementary in the early diagnosis, selecting treatment regimen, monitoring disease progression and evaluating prognosis [22, 126]. However, we should also keep the existing disadvantages of ctDNA in mind. Firstly, ctDNA detection was still quite low although KRAS mutations were quite high in pancreatic cancer tissue. The mechanism of ctDNA release and degeneration was still poorly understood [101]. Secondly, the ctDNA detection process hasn't been totally standardized at present. Only when standardized specimen preparation, detection technology and data analysis were carried out, could the ctDNA facilitate routine clinical decision. Thirdly, since there are only few targeted drugs for pancreatic cancer, we have no corresponding treatment regimen when ctDNA predicted early relapse. Early detection of disease may not prolong survival time or improve life quality. In contrast, this may bring extra psychological pressure [22]. Although there are some disadvantages, ctDNA could still play as a powerful weapon in clinical trials about prognosis, acquired drug resistance and treatment response, which would promote diagnosis and treatment on pancreatic cancer. CTCs, as viable and intact cells, are very tantalizing approach to perform biological studies, such as invasion, metastasis, and drug resistance both in vitro and in vivo.
In conclusion, CTCs are preferentially used for tumor biological studies and ctDNA is feasible for clinical research with great potential in translations medicine and precision medicine. The development of liquid biopsy is sure to provide essential information for clinical managements and prolong clinical outcomes in pancreatic cancer eventually.
Acknowledgements
This work was supported by Science and Technology Commission of Shanghai Municipality (Grant number: 15ZR1432200)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61:69-90
2. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039-49
3. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
4. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:9-29
5. Long J, Luo GP, Xiao ZW, Liu ZQ, Guo M, Liu L. et al. Cancer statistics: current diagnosis and treatment of pancreatic cancer in Shanghai, China. Cancer letters. 2014;346:273-7
6. Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nature reviews Clinical oncology. 2013;10:472-84
7. Overman MJ, Modak J, Kopetz S, Murthy R, Yao JC, Hicks ME. et al. Use of research biopsies in clinical trials: are risks and benefits adequately discussed? Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:17-22
8. Best MG, Sol N, Zijl S, Reijneveld JC, Wesseling P, Wurdinger T. Liquid biopsies in patients with diffuse glioma. Acta neuropathologica. 2015;129:849-65
9. Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J. et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177-82
10. Hayes DF, Paoletti C. Circulating tumour cells: insights into tumour heterogeneity. Journal of internal medicine. 2013;274:137-43
11. Alix-Panabieres C, Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clinical chemistry. 2013;59:110-8
12. Ting DT, Wittner BS, Ligorio M, Vincent Jordan N, Shah AM, Miyamoto DT. et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell reports. 2014;8:1905-18
13. Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S. et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487:510-3
14. Kolostova K, Pinkas M, Jakabova A, Pospisilova E, Svobodova P, Spicka J. et al. Molecular characterization of circulating tumor cells in ovarian cancer. American journal of cancer research. 2016;6:973-80
15. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F. et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349-61
16. Pierga JY, Bidard FC, Mathiot C, Brain E, Delaloge S, Giachetti S. et al. Circulating tumor cell detection predicts early metastatic relapse after neoadjuvant chemotherapy in large operable and locally advanced breast cancer in a phase II randomized trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:7004-10
17. Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF. et al. Circulating tumor cells in patients with breast cancer dormancy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:8152-62
18. Zhu YY, Yuan Z. Pancreatic cancer stem cells. American journal of cancer research. 2015;5:894-906
19. Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6:10697-711
20. Ignatiadis M, Lee M, Jeffrey SS. Circulating Tumor Cells and Circulating Tumor DNA: Challenges and Opportunities on the Path to Clinical Utility. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:4786-800
21. Nagrath S, Jack RM, Sahai V, Simeone DM. Opportunities and Challenges for Pancreatic Circulating Tumor Cells. Gastroenterology. 2016;151:412-26
22. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Science translational medicine. 2014;6:224ra24
23. Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nature reviews Cancer. 2011;11:426-37
24. Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clinical chemistry. 2015;61:112-23
25. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:579-86
26. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-6
27. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110-22
28. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399-405
29. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495-501
30. Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M. et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer discovery. 2015;5:1040-8
31. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C. et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:6897-904
32. Bidard FC, Huguet F, Louvet C, Mineur L, Bouche O, Chibaudel B. et al. Circulating tumor cells in locally advanced pancreatic adenocarcinoma: the ancillary CirCe 07 study to the LAP 07 trial. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2013;24:2057-61
33. Jiao LR, Apostolopoulos C, Jacob J, Szydlo R, Johnson N, Tsim N. et al. Unique localization of circulating tumor cells in patients with hepatic metastases. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:6160-5
34. Komar G, Kauhanen S, Liukko K, Seppanen M, Kajander S, Ovaska J. et al. Decreased blood flow with increased metabolic activity: a novel sign of pancreatic tumor aggressiveness. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:5511-7
35. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-90
36. Gao Y, Zhu Y, Zhang Z, Zhang C, Huang X, Yuan Z. Clinical significance of pancreatic circulating tumor cells using combined negative enrichment and immunostaining-fluorescence in situ hybridization. Journal of experimental & clinical cancer research: CR. 2016;35:66
37. Khoja L, Lorigan P, Dive C, Keilholz U, Fusi A. Circulating tumour cells as tumour biomarkers in melanoma: detection methods and clinical relevance. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2015;26:33-9
38. Imamura T, Komatsu S, Ichikawa D, Kawaguchi T, Miyamae M, Okajima W. et al. Liquid biopsy in patients with pancreatic cancer: Circulating tumor cells and cell-free nucleic acids. World J Gastroenterol. 2016;22:5627-41
39. Kulemann B, Liss AS, Warshaw AL, Seifert S, Bronsert P, Glatz T. et al. KRAS mutations in pancreatic circulating tumor cells: a pilot study. Tumour Biol. 2016;37:7547-54
40. Poruk KE, Valero V 3rd, Saunders T, Blackford AL, Griffin JF, Poling J. et al. Circulating Tumor Cell Phenotype Predicts Recurrence and Survival in Pancreatic Adenocarcinoma. Ann Surg. 2016
41. Bobek V, Gurlich R, Eliasova P, Kolostova K. Circulating tumor cells in pancreatic cancer patients: enrichment and cultivation. World J Gastroenterol. 2014;20:17163-70
42. Kulemann B, Pitman MB, Liss AS, Valsangkar N, Fernandez-Del Castillo C, Lillemoe KD. et al. Circulating tumor cells found in patients with localized and advanced pancreatic cancer. Pancreas. 2015;44:547-50
43. Kulemann B, Liss AS, Warshaw AL, Seifert S, Bronsert P, Glatz T. et al. KRAS mutations in pancreatic circulating tumor cells: a pilot study. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015
44. Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L. et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Science translational medicine. 2010;2:25ra3
45. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L. et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235-9
46. Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA. et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18392-7
47. Alix-Panabieres C, Pantel K. Challenges in circulating tumour cell research. Nature reviews Cancer. 2014;14:623-31
48. Pantel K, Speicher MR. The biology of circulating tumor cells. Oncogene. 2015
49. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC. et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216-20
50. Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT. et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349:1351-6
51. Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F. et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nature medicine. 2014;20:897-903
52. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:2140-1
53. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B. et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114-7
54. Fritz S, Klauss M, Bergmann F, Hackert T, Hartwig W, Strobel O. et al. Small (Sendai negative) branch-duct IPMNs: not harmless. Annals of surgery. 2012;256:313-20
55. Rhim AD, Thege FI, Santana SM, Lannin TB, Saha TN, Tsai S. et al. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology. 2014;146:647-51
56. Ilie M, Hofman V, Long-Mira E, Selva E, Vignaud JM, Padovani B. et al. "Sentinel" circulating tumor cells allow early diagnosis of lung cancer in patients with chronic obstructive pulmonary disease. PloS one. 2014;9:e111597
57. Z'Graggen K, Centeno BA, Fernandez-del Castillo C, Jimenez RE, Werner J, Warshaw AL. Biological implications of tumor cells in blood and bone marrow of pancreatic cancer patients. Surgery. 2001;129:537-46
58. Kurihara T, Itoi T, Sofuni A, Itokawa F, Tsuchiya T, Tsuji S. et al. Detection of circulating tumor cells in patients with pancreatic cancer: a preliminary result. Journal of hepato-biliary-pancreatic surgery. 2008;15:189-95
59. Zhou J, Hu L, Yu Z, Zheng J, Yang D, Bouvet M. et al. Marker expression in circulating cancer cells of pancreatic cancer patients. The Journal of surgical research. 2011;171:631-6
60. Khoja L, Backen A, Sloane R, Menasce L, Ryder D, Krebs M. et al. A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker. British journal of cancer. 2012;106:508-16
61. Sheng W, Ogunwobi OO, Chen T, Zhang J, George TJ, Liu C. et al. Capture, release and culture of circulating tumor cells from pancreatic cancer patients using an enhanced mixing chip. Lab on a chip. 2014;14:89-98
62. Cauley CE, Pitman MB, Zhou J, Perkins J, Kuleman B, Liss AS. et al. Circulating Epithelial Cells in Patients with Pancreatic Lesions: Clinical and Pathologic Findings. Journal of the American College of Surgeons. 2015;221:699-707
63. Zhang Y, Wang F, Ning N, Chen Q, Yang Z, Guo Y. et al. Patterns of circulating tumor cells identified by CEP8, CK and CD45 in pancreatic cancer. International journal of cancer Journal international du cancer. 2015;136:1228-33
64. Yamada T, Nakamori S, Ohzato H, Oshima S, Aoki T, Higaki N. et al. Detection of K-ras gene mutations in plasma DNA of patients with pancreatic adenocarcinoma: correlation with clinicopathological features. Clinical cancer research: an official journal of the American Association for Cancer Research. 1998;4:1527-32
65. Castells A, Puig P, Mora J, Boadas J, Boix L, Urgell E. et al. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: diagnostic utility and prognostic significance. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1999;17:578-84
66. Dianxu F, Shengdao Z, Tianquan H, Yu J, Ruoqing L, Zurong Y. et al. A prospective study of detection of pancreatic carcinoma by combined plasma K-ras mutations and serum CA19-9 analysis. Pancreas. 2002;25:336-41
67. Uemura T, Hibi K, Kaneko T, Takeda S, Inoue S, Okochi O. et al. Detection of K-ras mutations in the plasma DNA of pancreatic cancer patients. Journal of gastroenterology. 2004;39:56-60
68. Marchese R, Muleti A, Pasqualetti P, Bucci B, Stigliano A, Brunetti E. et al. Low correspondence between K-ras mutations in pancreatic cancer tissue and detection of K-ras mutations in circulating DNA. Pancreas. 2006;32:171-7
69. Dabritz J, Preston R, Hanfler J, Oettle H. Follow-up study of K-ras mutations in the plasma of patients with pancreatic cancer: correlation with clinical features and carbohydrate antigen 19-9. Pancreas. 2009;38:534-41
70. Chen H, Tu H, Meng ZQ, Chen Z, Wang P, Liu LM. K-ras mutational status predicts poor prognosis in unresectable pancreatic cancer. European journal of surgical oncology: the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 2010;36:657-62
71. Earl J, Garcia-Nieto S, Martinez-Avila JC, Montans J, Sanjuanbenito A, Rodriguez-Garrote M. et al. Circulating tumor cells (Ctc) and kras mutant circulating free Dna (cfdna) detection in peripheral blood as biomarkers in patients diagnosed with exocrine pancreatic cancer. BMC cancer. 2015;15:797
72. Takai E, Totoki Y, Nakamura H, Morizane C, Nara S, Hama N. et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Scientific reports. 2015;5:18425
73. Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT. et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nature communications. 2015;6:7686
74. Kinugasa H, Nouso K, Miyahara K, Morimoto Y, Dohi C, Tsutsumi K. et al. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer. 2015;121:2271-80
75. Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen-Ottosen Berry K, Gilje B. et al. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Molecular oncology. 2016;10:635-43
76. Paulson AS, Tran Cao HS, Tempero MA, Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013;144:1316-26
77. Cen P, Ni X, Yang J, Graham DY, Li M. Circulating tumor cells in the diagnosis and management of pancreatic cancer. Biochimica et biophysica acta. 2012;1826:350-6
78. Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV. et al. Detection of mutations in EGFR in circulating lung-cancer cells. The New England journal of medicine. 2008;359:366-77
79. Schiller JH. Noninvasive monitoring of tumors. The New England journal of medicine. 2008;359:418-20
80. Smerage JB, Barlow WE, Hortobagyi GN, Winer EP, Leyland-Jones B, Srkalovic G. et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:3483-9
81. Tjensvoll K, Nordgard O, Smaaland R. Circulating tumor cells in pancreatic cancer patients: methods of detection and clinical implications. International journal of cancer Journal international du cancer. 2014;134:1-8
82. Paterlini-Brechot P, Benali NL. Circulating tumor cells (CTC) detection: clinical impact and future directions. Cancer letters. 2007;253:180-204
83. Gall TM, Frampton AE, Krell J, Jacob J, Stebbing J, Jiao LR. Is the detection of circulating tumor cells in locally advanced pancreatic cancer a useful prognostic marker? Expert review of molecular diagnostics. 2013;13:793-6
84. de Albuquerque A, Kubisch I, Breier G, Stamminger G, Fersis N, Eichler A. et al. Multimarker gene analysis of circulating tumor cells in pancreatic cancer patients: a feasibility study. Oncology. 2012;82:3-10
85. Han L, Chen W, Zhao Q. Prognostic value of circulating tumor cells in patients with pancreatic cancer: a meta-analysis. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:2473-80
86. de Wit S, van Dalum G, Lenferink AT, Tibbe AG, Hiltermann TJ, Groen HJ. et al. The detection of EpCAM(+) and EpCAM(-) circulating tumor cells. Scientific reports. 2015;5:12270
87. Kling J. Beyond counting tumor cells. Nature biotechnology. 2012;30:578-80
88. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
89. Costello E, Greenhalf W, Neoptolemos JP. New biomarkers and targets in pancreatic cancer and their application to treatment. Nature reviews Gastroenterology & hepatology. 2012;9:435-44
90. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546-58
91. Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L. et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315-26
92. Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501:355-64
93. Katoh M. WNT signaling pathway and stem cell signaling network. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:4042-5
94. Nanus DM, Giannakakou P. CANCER. Cancer therapies that are gone with the Wnt. Science. 2015;349:1283-4
95. Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM. et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer research. 2013;73:2965-75
96. Li M, Wang H, Huang T, Wang J, Ding Y, Li Z. et al. TAB2 scaffolds TAK1 and NLK in repressing canonical Wnt signaling. The Journal of biological chemistry. 2010;285:13397-404
97. Plaks V, Koopman CD, Werb Z. Cancer. Circulating tumor cells. Science. 2013;341:1186-8
98. Hou JM, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK. et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:525-32
99. Molnar B, Ladanyi A, Tanko L, Sreter L, Tulassay Z. Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Clinical cancer research: an official journal of the American Association for Cancer Research. 2001;7:4080-5
100. Aktary Z, Pasdar M. Plakoglobin: role in tumorigenesis and metastasis. International journal of cell biology. 2012;2012:189521
101. Riva F, Dronov OI, Khomenko DI, Huguet F, Louvet C, Mariani P. et al. Clinical applications of circulating tumor DNA and circulating tumor cells in pancreatic cancer. Molecular oncology. 2016;10:481-93
102. Chari ST, Kelly K, Hollingsworth MA, Thayer SP, Ahlquist DA, Andersen DK. et al. Early detection of sporadic pancreatic cancer: summative review. Pancreas. 2015;44:693-712
103. Okano K, Suzuki Y. Strategies for early detection of resectable pancreatic cancer. World journal of gastroenterology. 2014;20:11230-40
104. Ghaneh P, Costello E, Neoptolemos JP. Biology and management of pancreatic cancer. Gut. 2007;56:1134-52
105. Gao F, Liu QC, Zhang S, Zhuang ZH, Lin CZ, Lin XH. PRSS1 intron mutations in patients with pancreatic cancer and chronic pancreatitis. Molecular medicine reports. 2012;5:449-51
106. Schubert S, Traub F, Brakensiek K, von Kopylow K, Marohn B, Maelzer M. et al. CFTR, SPINK1, PRSS1, and CTRC mutations are not associated with pancreatic cancer in German patients. Pancreas. 2014;43:1078-82
107. Saxena N, Lahiri SS, Hambarde S, Tripathi RP. RAS: target for cancer therapy. Cancer investigation. 2008;26:948-55
108. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer cell. 2014;25:304-17
109. Sundaresan TK, Sequist LV, Heymach JV, Riely GJ, Janne PA, Koch WH. et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015
110. Beaver JA, Jelovac D, Balukrishna S, Cochran RL, Croessmann S, Zabransky DJ. et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:2643-50
111. Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer-a survey. Biochimica et biophysica acta. 2007;1775:181-232
112. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M. et al. Circulating mutant DNA to assess tumor dynamics. Nature medicine. 2008;14:985-90
113. Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ. et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Science translational medicine. 2015;7:302ra133
114. Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK. et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63:318-48
115. Madic J, Kiialainen A, Bidard FC, Birzele F, Ramey G, Leroy Q. et al. Circulating tumor DNA and circulating tumor cells in metastatic triple negative breast cancer patients. International journal of cancer Journal international du cancer. 2015;136:2158-65
116. Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA. et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109-13
117. Silva JM, Silva J, Sanchez A, Garcia JM, Dominguez G, Provencio M. et al. Tumor DNA in plasma at diagnosis of breast cancer patients is a valuable predictor of disease-free survival. Clinical cancer research: an official journal of the American Association for Cancer Research. 2002;8:3761-6
118. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends in biochemical sciences. 2014;39:91-100
119. Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. British journal of cancer. 2014;111:817-22
120. Kinugasa H, Nouso K, Miyahara K, Morimoto Y, Dohi C, Tsutsumi K. et al. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer. 2015
121. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J. et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nature communications. 2015;6:6744
122. Oliveira C, Velho S, Moutinho C, Ferreira A, Preto A, Domingo E. et al. KRAS and BRAF oncogenic mutations in MSS colorectal carcinoma progression. Oncogene. 2007;26:158-63
123. Monticone M, Biollo E, Maffei M, Donadini A, Romeo F, Storlazzi CT. et al. Gene expression deregulation by KRAS G12D and G12V in a BRAF V600E context. Molecular cancer. 2008;7:92
124. Siravegna G, Bardelli A. Minimal residual disease in breast cancer: in blood veritas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:2505-7
125. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. The New England journal of medicine. 2013;368:1199-209
126. Saliou A, Bidard FC, Lantz O, Stern MH, Vincent-Salomon A, Proudhon C. et al. Circulating tumor DNA for triple-negative breast cancer diagnosis and treatment decisions. Expert review of molecular diagnostics. 2015:1-12
127. Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K. et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nature reviews Drug discovery. 2012;11:873-86
128. Nedaeinia R, Avan A, Manian M, Salehi R, Ghayour-Mobarhan M. EGFR as a potential target for the treatment of pancreatic cancer: dilemma and controversies. Current drug targets. 2014;15:1293-301
129. Weiner GJ. Building better monoclonal antibody-based therapeutics. Nature reviews Cancer. 2015;15:361-70
130. Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013;341:1192-8
131. Heinemann V, Vehling-Kaiser U, Waldschmidt D, Kettner E, Marten A, Winkelmann C. et al. Gemcitabine plus erlotinib followed by capecitabine versus capecitabine plus erlotinib followed by gemcitabine in advanced pancreatic cancer: final results of a randomised phase 3 trial of the 'Arbeitsgemeinschaft Internistische Onkologie' (AIO-PK0104). Gut. 2013;62:751-9
132. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S. et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960-6
133. Yang ZY, Yuan JQ, Di MY, Zheng DY, Chen JZ, Ding H. et al. Gemcitabine plus erlotinib for advanced pancreatic cancer: a systematic review with meta-analysis. PloS one. 2013;8:e57528
134. Cinar P, Tempero MA. Monoclonal antibodies and other targeted therapies for pancreatic cancer. Cancer J. 2012;18:653-64
135. Bruns CJ, Harbison MT, Davis DW, Portera CA, Tsan R, McConkey DJ. et al. Epidermal growth factor receptor blockade with C225 plus gemcitabine results in regression of human pancreatic carcinoma growing orthotopically in nude mice by antiangiogenic mechanisms. Clinical cancer research: an official journal of the American Association for Cancer Research. 2000;6:1936-48
136. Solorzano CC, Baker CH, Bruns CJ, Killion JJ, Ellis LM, Wood J. et al. Inhibition of growth and metastasis of human pancreatic cancer growing in nude mice by PTK 787/ZK222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases. Cancer biotherapy & radiopharmaceuticals. 2001;16:359-70
Author contact
![]() Corresponding author: Zhou Yuan, zhouyuan669com
Corresponding author: Zhou Yuan, zhouyuan669com