Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2016; 13(10):754-758. doi:10.7150/ijms.15847 This issue Cite
Research Paper
Role of CAPE on cardiomyocyte protection via connexin 43 regulation under hypoxia
Chien-Cheng Chen1*, Chan-Yen Kuo2*, Rong-Fu Chen3 ![]()
1. Department of Cardiology, Show Chwan Memorial Hospital, Changhua, Taiwan.
2. Graduate Institute of Systems Biology and Bioinformatics, National Central University, Chung-li, Taiwan, 32001, Republic of China.
3. Research Assistant Center, Show Chwan Health Care System, Changhua, Taiwan.
*These two authors equally contribute to this study.
Received 2016-4-14; Accepted 2016-8-22; Published 2016-9-20
Abstract
Background: Cardiomyocyte under hypoxia cause cell death or damage is associated with heart failure. Gap junction, such as connexin 43 play a role in regulation of heart function under hypoxia. Caffeic acid phenethyl ester (CAPE) has been reported as an active component of propolis, has antioxidative, anti-inflammatory antiproliferative and antineoplastic biological properties.
Aims: Connexin 43 appear to have a critical role in heart failure under hypoxia, there has been considerable interest in identifying the candidate component or compound to reduce cell death.
Methods: In this study, we used human cardiomyocyte as a cell model to study the role of connexin 43 in hypoxia- incubated human cardiomyocyte in absence or presence of CAPE treatment.
Results: Results showed that hypoxia induced connexin 43 expression, but not altered in connexin 40. Interestingly, CAPE attenuates hypoxia-caused connexin 43 down-regulation and cell death or cell growth inhibition.
Conclusion: We suggested that reduction of cell death in cardiomyocytes by CAPE is associated with an increase in connexin 43 expression.
Keywords: Caffeic acid phenethyl ester (CAPE), cardiomyocyte, hypoxia, connexin 43.
Introduction
Coronary artery disease (CAD) is one of major diseases causing heavy burden of many countries and people around the world [1]. Atherosclerosis is the main cause of CAD and involved in endothelial dysfunction and inflammation [2-4]. Furthermore, previous studies reported that exercise is a secondary prevention of CAD [5], and some reports indicated that exercise seems to be improved the endothelial function [6, 7]. Reactive oxygen species (ROS) and oxidative stress are important feature of cardiovascular diseases including atherosclerosis, hypertension, and congestive failure [8]. Similar result showed that ROS production is increased in mitochondria upon hypoxia, as well as, ischemic preconditioning (IPC) [9-11]. Hypoxia-inducible factor transcription factors (HIF) is upregulated upon hypoxia [12], and triggers the expression of genes involved in oxygen transport, glycolytic metabolism, cell death, cell survival, and other processes that can affect cell survival in ischemia [10].
Caffeic acid phenethyl ester (CAPE) has been reported that it is the major active element of propolis and has an anti- proliferative effect on tumor cells [13, 14]. The antioxidative activities of CAPE have been reported in vitro and in different biological systems [15, 16].
Gap junctions, which are specialized cell-cell contacts between cardiomyocytes in the heart, mediate the electrical coupling of cardiomyocytes and as such dictate the speed and direction of cardiac conduction [17]. Communication of nitric oxide signaling and gap junctions (connexins) are essential for normal vascular function [18]. Moreover, the gap junctions have been reported that participates in mediating various cell fates in vitro [19], however, the effect of hypoxia on human cardiomyocyte is still uncertain. Connexin 43 (Cx43) is a major structural protein found in the gap junctions of the ventricular myocardium and a major determinant of its electrical properties [20], as well as, it also has been considered as a major component of cardiomyocyte gap junctions, which regulate intercellular coupling, conduction velocity, and anisotropy [21, 22]. Additionally, Cx43-formed channels are involved in ischemia/reperfusion injury and ischemic preconditioning's protection in vitro [23], however, the role of Cx43 on human cardiomyocyte under hypoxia is still unclear. Previous report demonstrated that Cx43 is highly expressed in cardiomyocytes and is involved in IPC [23, 24]. Isolated cardiomyocytes from wild type mice can protect of IPC [25], but form heterozygous Cx43-deficient mice cannot [26]. An additional role of Cx43 in the mitochondria is considered as a critical role in the pathophysiology of myocardial ischaemia-reperfusion injury and regulation of reactive oxygen species during IPC [27]. Interestingly, replacement of Cx43 by Cx32 attenuates aortic endothelium-derived hyperpolarizing factor (AEDH)-mediated relaxation in mice [28]. Therefore, the role of Cx43 in regulation of mitochondrial function of cardiomyocytes under ischaemia or hypoxia is very important for us to know.
Materials and methods
Cell lines and cell culture
Human cardiomyocyte (HCM) (PromoCell GmbH, Heidelberg, Germany) were cultured at 37 ° C in T-25 flasks (Corning Glassworks, Corning, N.Y., USA) in Myocyte Growth Medium (PromoCell GmbH, Heidelberg, Germany) supplemented
with 0.05 ml/ml fetal calf serum, 0.5 ng/ml epidermal growth factor, 2 ng/ml basic fibroblast growth factor, and 5 µg/ml insulin in a 5% CO2/95% air atmosphere. The culture medium was replaced every 2 days. Once the cells reached 70-80% confluence, they were trypsinized and seeded on 6-well plastic dishes for the following experiments. Passage 3-9 HCMs were used in the experiment.
Western Blotting
Cells were extracted by ice-cold RIPA buffer (20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EGTA, 1 mM NaF, 2 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 1% dilution of Sigma protease cocktail, and 1% Triton X-100). Samples were centrifuged at 10,000 g for 15 min at 4 °C to yield cell lysates. Proteins were separated by 10% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoresed onto a nitrocellulose membrane. Immunoblotting was performed using specific primary antibodies and horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology, MA, USA), and peroxidase activity was assessed using an enhanced chemiluminescence kit (Perkin-Elmer Life Science, Boston, MA, USA). The intensities of the reactive bands were analyzed using ImageJ.
Establishment of hypoxic culture condition
Hypoxic conditions were assessed using a method previously described [29] with some modifications. Briefly, cells were grown on 6 well plastic dishes, in a hypoxia chamber and equilibrating for 30 minutes with humidified gas containing 1 % oxygen, 5 % CO2 and 94 % nitrogen (Hypoxic incubator APM-30D, Astec., Tokyo). The cell lines were maintained under hypoxic conditions for various time courses. Control cells were grown in normal oxygen conditions for the same duration.
Lactate dehydrogenase (LDH) release and cytotoxicity assay
The analysis was performed using the LDH Cytotoxicity Assay Kit (Pierce IL, USA) according to the manufacturer's instructions. Statistical significance for all the experiments was determined by performing the t test. Error bars are used to indicate the standard errors of the means and p values of < 0.01 were considered significant.
Results
Effect of hypoxia on connexin 43 and cell LDH release
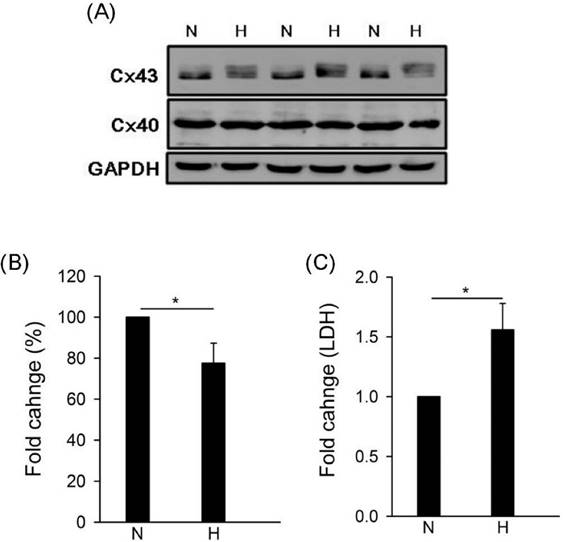
To study the effect of hypoxia on gap junction in human cardiomyocytes, we detected the expressions of connexin 43 (Cx43) and connexin 40 (Cx40) by western blotting under hypoxia. Results showed that hypoxia caused Cx43 down-regulation but no effect on Cx40 (Fig. 1A and B). Moreover, LDH is a biomarker for cell cytotoxicity and cytolysis. Loss of intracellular LDH and its release into the culture medium has been considered as an indicator of irreversible cell death via cell membrane damage [30]. In this study, results showed that hypoxia caused LDH significantly release (Fig. 1C).
Effect of hypoxia on gap junction expression and LDH release. Cells were incubated under hypoxia or normoxia treatment for 24h (western blotting) and 48hh (LDH). (A) Detection of connexin 43, 40, and GAPDH by western blotting with specific antibody. (B) The quantitative results on the level of specific protein (connexin 43) by ImageJ. All data are presented as the mean ± S.E.M. * p <0.05 as compared with normoxia (N). (C) Detection of LDH release. N: normoxia; H: hypoxia.
Effect of CAPE on connexin 43 expression and cell number under hypoxia in human cardiomyocyte
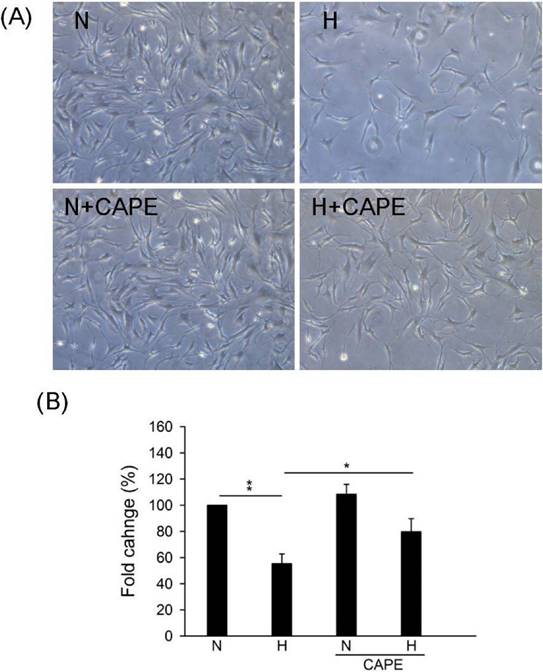
In our previous study, we suggested that CAPE controls ROS accumulation and cell death in cardiomyocyte [31]. However, the effect of CAPE on Cx43 in human cardiomyocyte is still uncertain. Results showed that 30 μM CAPE reversed the down-regulation under hypoxia in human cardiomyocyte (Fig. 2). We further speculated the role of CAPE on cell number under hypoxia, results showed that CAPE attenuated cell growth inhibition or death caused by hypoxia (Fig. 3). Therefore, we suggested that CAPE prevented human cardiomyocytes death via connexin 43 under hypoxia.
Effect of CAPE on connexin 43 expression (arrow) under hypoxia. Cells were incubated in presence or absence of 30 μM CAPE and under hypoxia or normoxia treatment for 24h. GAPDH is an internal control.

Effect of CAPE on cell survival under hypoxia. Cells were incubated in presence or absence of 30 μM CAPE and under hypoxia or normoxia treatment for 48h. (A) Morphological images were observed in the indicated conditions. (B) The quantitative results on the cell numbers. All data are presented as the mean ± S.E.M. * p <0.05; ** p <0.01 as compared with the corresponding group. N: Normoxia; N+C: Normoxia + 30 μM CAPE; H: Hypoxia; H+C: Hypoxia + 30 μM CAPE.
Discussion
Oxidative stress-induced apoptotic signaling can cause several pathological conditions, including the development and progression of heart disease, which are a consequence of the increases ROS or decreases in antioxidants, as well as a disruption in the intracellular redox homeostasis [32-34]. Our previous study demonstrated that CAPE reverses hypoxia-induced LDH release and cell death in human cardiomyocytes, as well as ROS scavenger, Tiron also prevents H2O2 induces LDH release and cytotoxicity [31]. However, the role of CAPE on gap junction in human cardiomyocytes is still unclear. Therefore, we detected the expression of connexin 43 under hypoxia in the absence or presence of CAPE. Results showed that CAPE prevented hypoxia-caused connexin 43 down-regulation, however, hypoxia did not alter connexin 40 expression. According to previous reports, connexin 43 plays a more critical role than other connexins in the atrial and ventricular working myocardium and in the distal Purkinje system [35, 36]. Furthermore, connexin43 has been reported that it located in the nuclear membrane and in subsarcolemmal mitochondria [37]. Boengler et al. reported that ischemic preconditioning causes increasing in levels of mitochondrial connexin43 [38]. Mitochondria are a major source of ROS, which in excessive amounts induce oxidative stress and promote cell death under hypoxic conditions. However, at physiological levels, ROS function as “redox messengers” in intracellular signaling [39]. Therefore, we suggested that CAPE controls cardiomyocytes death or survival via connexin 43-ROS dependent pathway, however, we need more evidence to verify it.
Acknowledgements
This study was supported by grants 105-2914-I-008 -008 -A1 from Ministry of Science and Technology, Taiwan.
Competing Interests
None of the authors has any potential financial competing interest related to this paper.
References
1. Scott J. Pathophysiology and biochemistry of cardiovascular disease. Opin Genet Dev. 2004;14:271-9
2. Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation. 2004;109:II27-33
3. Libby P, Aikawa M, Jain MK. Vascular endothelium and atherosclerosis. Handbook Exp Pharmacol. 2006:285-306
4. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685-95
5. Lavie CJ, Thomas RJ, Squires RW, Allison TG, Milani RV. Exercise training and cardiac rehabilitation in primary and secondary prevention of coronary heart disease. Mayo Clin Proc. 2009;84:373-83
6. Gielen S, Schuler G, Hambrecht R. Exercise training in coronary artery disease and coronary vasomotion. Circulation. 2001;103:E1-6
7. Ribeiro F, Alves AJ, Duarte JA, Oliveira J. Is exercise training an effective therapy targeting endothelial dysfunction and vascular wall inflammation? Int J Cardiol. 2010;141:214-21
8. Sugamura K, Keaney JF Jr. Reactive oxygen species in cardiovascular disease. Free Radical Biol Med. 2011;51:978-92
9. Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem. 1998;273:11619-24
10. Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008;15:686-90
11. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524-37
12. Mazure NM, Brahimi-Horn MC, Berta MA, Benizri E, Bilton RL, Dayan F. et al. HIF-1: master and commander of the hypoxic world. A pharmacological approach to its regulation by siRNAs. Biochem Pharmacol. 2004;68:971-80
13. Kuo HC, Kuo WH, Lee YJ, Lin WL, Chou FP, Tseng TH. Inhibitory effect of caffeic acid phenethyl ester on the growth of C6 glioma cells in vitro and in vivo. Cancer Lett. 2006;234:199-208
14. Kuo HC, Kuo WH, Lee YJ, Wang CJ, Tseng TH. Enhancement of caffeic acid phenethyl ester on all-trans retinoic acid-induced differentiation in human leukemia HL-60 cells. Toxicol Appl Pharm. 2006;216:80-8
15. Hsu LY, Lin CF, Hsu WC, Hsu WL, Chang TC. Evaluation of polyphenolic acid esters as potential antioxidants. Biol Pharm Bull. 2005;28:1211-5
16. Jia CH, Wang XY, Qi JF, Hong ST, Lee KT. Antioxidant Properties of Caffeic acid Phenethyl Ester and 4-Vinylcatechol in Stripped Soybean Oil. J Food Sci. 2015
17. Hesketh GG, Van Eyk JE, Tomaselli GF. Mechanisms of gap junction traffic in health and disease. J Cardiovasc Pharm. 2009;54:263-72
18. Looft-Wilson RC, Billaud M, Johnstone SR, Straub AC, Isakson BE. Interaction between nitric oxide signaling and gap junctions: Effects on vascular function. BBA-Biomembranes. 2011
19. Vinken M, Vanhaecke T, Papeleu P, Snykers S, Henkens T, Rogiers V. Connexins and their channels in cell growth and cell death. Cell Signal. 2006;18:592-600
20. Wu X, Huang W, Luo G, Alain LA. Hypoxia induces connexin 43 dysregulation by modulating matrix metalloproteinases via MAPK signaling. Mol Cell Biochem. 2013;384:155-62
21. van Veen AA, van Rijen HV, Opthof T. Cardiac gap junction channels: modulation of expression and channel properties. Cardiovasc Res. 2001;51:217-29
22. Cabo C, Boyden PA. Extracellular space attenuates the effect of gap junctional remodeling on wave propagation: a computational study. Biophys J. 2009;96:3092-101
23. Schulz R, Heusch G. Connexin 43 and ischemic preconditioning. Cardiovasc Res. 2004;62:335-44
24. Li X, Heinzel FR, Boengler K, Schulz R, Heusch G. Role of connexin 43 in ischemic preconditioning does not involve intercellular communication through gap junctions. J Mol Cell Cardiol. 2004;36:161-3
25. Schwanke U, Konietzka I, Duschin A, Li X, Schulz R, Heusch G. No ischemic preconditioning in heterozygous connexin43-deficient mice. Am J Physiol-Heart Circ Physiol. 2002;283:H1740-2
26. Li G, Whittaker P, Yao M, Kloner RA, Przyklenk K. The gap junction uncoupler heptanol abrogates infarct size reduction with preconditioning in mouse hearts. Cardiovascu Pathol. 2002;11:158-65
27. Ruiz-Meana M, Rodriguez-Sinovas A, Cabestrero A, Boengler K, Heusch G, Garcia-Dorado D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2008;77:325-33
28. Lopez D, Rodriguez-Sinovas A, Agullo E, Garcia A, Sanchez JA, Garcia-Dorado D. Replacement of connexin 43 by connexin 32 in a knock-in mice model attenuates aortic endothelium-derived hyperpolarizing factor-mediated relaxation. Exp Physiol. 2009;94:1088-97
29. van Dyck L, Neupert W, Langer T. The ATP-dependent PIM1 protease is required for the expression of intron-containing genes in mitochondria. Genes Dev. 1998;12:1515-24
30. Fotakis G, Timbrell JA. In vitro cytotoxicity assays: comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol Lett. 2006;160:171-7
31. Huan-Nung Chao C-HL, Chien-Cheng Chen, Chun-Yen Huang, Chan-Yen Kuo. Caffeic acid phenethyl ester (CAPE) reduces LDH release and cell cytotoxicity in cardiomyocyte. Am J Pharm Pharmacol. 2015;3:5
32. Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343-74
33. Baines CP. The cardiac mitochondrion: nexus of stress. Annu Rev Physiol. 2010;72:61-80
34. Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19-44
35. Plum A, Hallas G, Magin T, Dombrowski F, Hagendorff A, Schumacher B. et al. Unique and shared functions of different connexins in mice. Curr Biol. 2000;10:1083-91
36. Verheule S, Kaese S. Connexin diversity in the heart: insights from transgenic mouse models. Front Pharmacol. 2013;4:81
37. Rodriguez-Sinovas A, Cabestrero A, Lopez D, Torre I, Morente M, Abellan A. et al. The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog Biophys Mol Biol. 2007;94:219-32
38. Boengler K, Dodoni G, Rodriguez-Sinovas A, Cabestrero A, Ruiz-Meana M, Gres P. et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res. 2005;67:234-44
39. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749-62
Author contact
![]() Corresponding author: Dr. Rong-Fu Chen, Research Assistant Center, Show Chwan Health Care System, 5th Floor, 5th Medical Building, No. 542, Sec 1 Chung-Shan Rd., Changhua. 500, Taiwan. Phone: 886-4-7813888 ext. 72691, Fax: 886-4-7811426. E-mail: den2.th1com.
Corresponding author: Dr. Rong-Fu Chen, Research Assistant Center, Show Chwan Health Care System, 5th Floor, 5th Medical Building, No. 542, Sec 1 Chung-Shan Rd., Changhua. 500, Taiwan. Phone: 886-4-7813888 ext. 72691, Fax: 886-4-7811426. E-mail: den2.th1com.