Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2016; 13(9):653-663. doi:10.7150/ijms.16232 This issue Cite
Research Paper
Preventative Effects of Sodium Alginate on Indomethacin-induced Small-intestinal Injury in Mice
Sayo Horibe1, Toshihito Tanahashi1,2, Shoji Kawauchi3, Shigeto Mizuno1,4, Yoshiyuki Rikitake1,5 ![]()
1. Department of Medical Pharmaceutics, Kobe Pharmaceutical University, Kobe 658-8558, Japan;
2. Division of Gastroenterology, Department of Internal Medicine, Kobe University Graduate School of Medicine, Kobe 650-0017, Japan
3. Educational Center for Clinical Pharmacy, Kobe Pharmaceutical University, Kobe 658-8558, Japan;
4. Endoscopy Department, Kindai University Nara Hospital, Ikoma 630-0293, Japan;
5. Division of Signal Transduction, Department of Biochemistry and Molecular Biology, Kobe University Graduate School of Medicine, Kobe 650-0017, Japan.
Received 2016-5-19; Accepted 2016-7-15; Published 2016-8-1
Abstract
Recent advances in diagnostic technologies have revealed that nonsteroidal anti-inflammatory drugs (NSAIDs) can cause serious mucosal injury in the upper and lower gastrointestinal tract (including the small intestine). A drug to treat NSAID-induced small-intestinal injury (SII) is lacking. Sodium alginate is a soluble dietary fiber extracted from brown seaweed and its solution has been used as a hemostatic agent to treat gastrointestinal bleeding due to gastric ulcers. Whether sodium alginate has therapeutic effects on NSAID-induced SII and its mechanism of action are not known. Here, we investigated if administration of two forms (high-molecular-weight (HMW) and low-molecular-weight (LMW)) of sodium alginate could ameliorate indomethacin-induced SII. Pretreatment with HMW sodium alginate or LMW sodium alginate before indomethacin administration improved ulceration and the resultant intestinal shortening was associated with reduced histological severity of mucosal injury and ameliorated mRNA expression of inflammation-related molecules in the small intestine. We found that mRNAs of secretory Muc2 and membrane-associated Muc1, Muc3 and Muc4 were expressed in the small intestine. mRNA expression of Muc1-4 was increased in indomethacin-induced SII, and these increases were prevented by sodium alginate. Thus, administration of sodium alginate could be a therapeutic approach to prevent indomethacin-induced SII.
Keywords: gene expression, mucin, nonsteroidal anti-inflammatory drugs, small intestine, sodium alginate.
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) such as indomethacin are used against inflammation and pain, but serious adverse effects on the gastrointestinal tract (GIT) and kidney can limit use. They can cause serious mucosal injury to the upper GIT [1], but recent advances in diagnostic technologies have revealed that they can cause serious mucosal injury to the small intestine [2, 3]. In contrast to the rationale for preventing damage to the upper GIT by acid suppression using proton pump inhibitors, such a strategy cannot prevent NSAID-induced small-intestinal injury (SII) [4, 5] and can exacerbate such injury in rats by induction of dysbiosis [6]. A drug to treat NSAID-induced SII is lacking.
Several factors have been shown to be involved in NSAID-induced SII [5, 7-9], but the mechanisms of NSAID-induced SII are complicated [5]. NSAIDs hamper synthesis of prostaglandin E2 by inhibiting cyclooxygenase (COX), thereby enhancing small-intestinal motility and reducing mucus secretion [10], which results in minute injury that allows intestinal bacteria to invade the mucosa. Lipopolysaccharide produced by intestinal bacteria elicits production of inducible nitric oxide synthase (iNOS), which causes production of nitric oxide and reactive oxygen species. These gases induce neutrophil activation and thereby COX-2 induction as well as production of inflammatory cytokines and mediators, which causes progression of intestinal inflammation [11]. Thus, a NSAID-impaired mucosal defense system causes erosion and ulcers within the small intestine [12, 13].
The surface of the intestinal mucosa is covered with mucins and glycoproteins that constitute a “mucous barrier” between the intestinal epithelial wall and external environment. Healthy mucosa can be protected from adverse conditions by mucous layers that contain mucins, but in injured mucosa, different mucins from those in healthy mucosa are expressed [14].
Sodium alginate is a soluble dietary fiber extracted from brown seaweed. It is a heteropolymer of D-mannuronic acid and L-guluronic acid [15]. The molecular weight of sodium alginate depends on by hydrolysis condition in its production. A solution of high-molecular-weight (HMW; 32-250 kD) sodium alginate is used as a hemostatic agent to treat GIT bleeding due to gastric and duodenal ulcers, erosion of gastric mucosa, and reflux esophagitis [16]. Because of its high viscosity, taking a sufficient amount of HMW sodium alginate to improve constipation and lower cholesterol levels in blood is difficult. Therefore, low-molecular-weight (LMW; average 50 kD) sodium alginate is available in Japan as a dietary supplement that promotes cholesterol excretion, relieves constipation, and suppresses carbohydrate absorption.
Our research team has developed a mouse model of indomethacin-induced SII to: (i) identify the underlying mechanism of action; and (ii) find a drug to prevent SII [17]. Here, we examined if and how HMW sodium alginate and LMW sodium alginate can prevent indomethacin-induced SII in mice.
Material and Methods
Indomethacin-induced SII
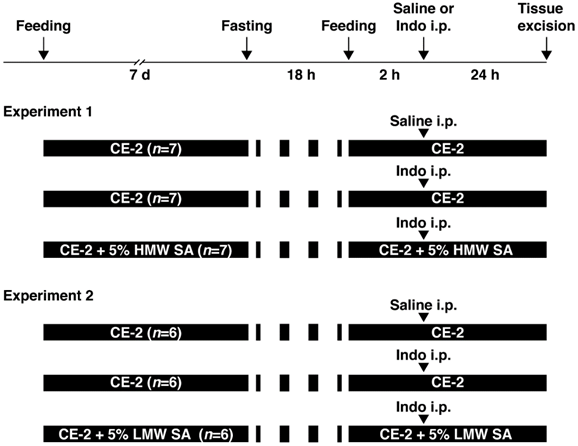
Animal experiments were approved by the Animal Care Committee of Kobe Pharmaceutical University (Kobe, Japan). Indomethacin-induced SII was induced as described previously [17]. Male C57BL/6 mice (7 weeks) were purchased from CLEA Japan (Shizuoka, Japan). They were provided with powdered CE-2 (CLEA Japan) and water ad libitum. Then, they were provided with regular CE-2 (n=6-7) or CE-2 premixed with 5% (w/v) HMW (n=7) or LMW sodium alginate (n=6), kind gifts from Kaigen Pharma (Osaka, Japan), for 7 days. To induce SII, mice were fasted for 18 h and then indomethacin (Sigma-Aldrich, Saint Louis, MO, USA) in physiologic (0.9%) saline with Tween 80 was administered (10 mg/5 mL/kg, s.c.). Twenty-four hours after indomethacin administration, mice were anesthetized (isoflurane inhalation) and small intestines excised (Figure 1).
Study protocol. Two experiments were conducted to examine the effects of high-molecular-weight sodium alginate (HMW SA) and low-molecular-weight sodium alginate (LMW SA) on indomethacin-induced small-intestinal injury in mice. i.p.: intraperitoneal injection.
Histology
The small intestine was excised and opened along the anti-mesenteric attachment. SII was evaluated by noting the area of macroscopically visible ulcers and length of the small intestine. Injured segments of the small intestine were trimmed and fixed in neutral-buffered 10% formalin solution, and then processed as tissue blocks for evaluation by hematoxylin and eosin staining. Injury was assessed by a scoring system for histological damage comprising four factors: width of ulceration (0 to 2); depth of the lesion (0 to 4); degree of inflammatory infiltration (0 to 3); thrombi (0 and 1) [18]. Thus, the range of histological score was from 0 to 10. Two scorers blinded to treatment groups undertook scoring of each section individually.
Additional paraffin-embedded sections (4 µm) were subjected to periodic acid-Schiff (PAS) staining. PAS-stained cells in the apical surface and basal-crypt layers of small-intestinal ulcers were scored. Staining was classified into one of four categories: “high” (≈90-100% of cells, score = 3), “medium” (25-90%, score = 2), “low” (1-25%, score = 1) and “virtually none” (<1%, score = 0) [19]. Two investigators blinded to treatment groups scored each section individually. Histology scores were assessed by calculating the Cohen's kappa coefficient (CKC) with SPSS v22.0 (IBM, Armonk, NY, USA).
Quantitative real-time polymerase chain reaction (PCR)
Total RNA was extracted from the small intestine using TRIzol™ Reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer instructions. RNA quantity was measured using a ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Two-hundred nanograms of total RNA were subjected to reverse transcription. Purified RNA was reverse-transcribed with a ReverTra Ace® qPCR RT kit (Toyobo, Osaka, Japan). PCR amplification was done on a Thermal Cycler Dice Real Time System (TaKaRaBio, Otsu, Japan) with Thunderbird® SYBR® Green PCR Master Mix (Toyobo). PCR conditions were 40 cycles of 15 s at 95°C for denaturation and 60 s at 60°C for annealing and extension. Primer sequences are shown in Table 1. Relative expression was calculated using the delta-delta Ct method with β-actin used as a reference gene [20].
Primer sequences.
| Gene | NM_ID | Product size (bp) | Sequence (5'→3') | Exon | |
|---|---|---|---|---|---|
| Tnf | NM_013693.2 | 103 | Fw | CCACCACGCTCTTCTGTCTAC | Exon 1 |
| Rv | AGGGTCTGGGCCATAGAACT | Exon 2 | |||
| Il1b | NM_008361.3 | 152 | Fw | CAACCAACAAGTGATATTCTCCATG | Exons 5-6 |
| Rv | GATCCACACTCTCCAGCTGCA | Exons 6-7 | |||
| Il6 | NM_031168.1 | 102 | Fw | CTGGAGTCACAGAAGGAGTGG | Exon 5 |
| Rv | GGTTTGCCGAGTAGATCTCAA | Exon 5 | |||
| Nos2 | NM_010927.3 | 95 | Fw | CAGCTGGGCTGTACAAACCTT | Exon 17 |
| Rv | CATTGGAAGTGAAGCGTTTCG | Exon 18 | |||
| Ptgs2 | NM_011198.3 | 129 | Fw | GGTGCCTGGTCTGATGATGTATG | Exon 7 |
| Rv | ATGAGTATGAGTCTGCTGGTTTGG | Exons 7-8 | |||
| Actb | NM_009898.2 | 138 | Fw | AGAGGGAAATCGTGCGTGAC | Exon 4 |
| Rv | CAATAGTGATGACCTGGCCGT | Exon 4 | |||
| Muc1 | NM_013605.1 | 165 | Fw | TTCTTGCCCTTCCAAGTGAG | Exon 1 |
| Rv | GTCTGAGTTGCTGCTGTGGA | Exon 2 | |||
| Muc2 | NM_023566.3 | 166 | Fw | TCTGGCATCAACATTGTGGT | Exon 2 |
| Rv | CACTGGGCAGGATACAGTCA | Exon 3 | |||
| Muc3 | XM_355711.9 | 153 | Fw | GCTGAAGTAACCACCACTGCT | Exon 1 |
| Rv | GCACTTGTCACCTGTCCAGA | Exon 2 | |||
| Muc4 | NM_080457.3 | 147 | Fw | GAGAGTTCCCTGGCTGTGTC | Exon 1 |
| Rv | GATGAGGTCGATGCTTGTGA | Exon 2 | |||
| Muc5ac | NM_010844.1 | 83 | Fw | GCTGGAGTTGGACACCAAATA | Exon 5 |
| Rv | GGAACTCGTTGGATTTTGGA | Exon 6 | |||
| Muc5b | NM_028801.2 | 121 | Fw | AGAAACTGGAGCTGGGCTCT | Exon 1 |
| Rv | TCTGACTGTCTCCGGTGAGTT | Exon 2 | |||
| Muc6 | NM_181729.2 | 62 | Fw | CTGCTGTTGCTGCTCTTCAG | Exon 1 |
| Rv | ATACTCATGGCCGTCAAAGG | Exon 3 |
Statistical analyses
Data are the mean ± standard error (SEM) of independent determinations for each experiment. Significance was analyzed using Student's t-test, and comparisons of more than two groups were evaluated with one-way analysis of variance followed by the Tukey test as appropriate with SPSS v22.0. A value of P<0.05 was considered significant.
Results
Reduction of the histological severity of indomethacin-induced SII in mice by HMW sodium alginate
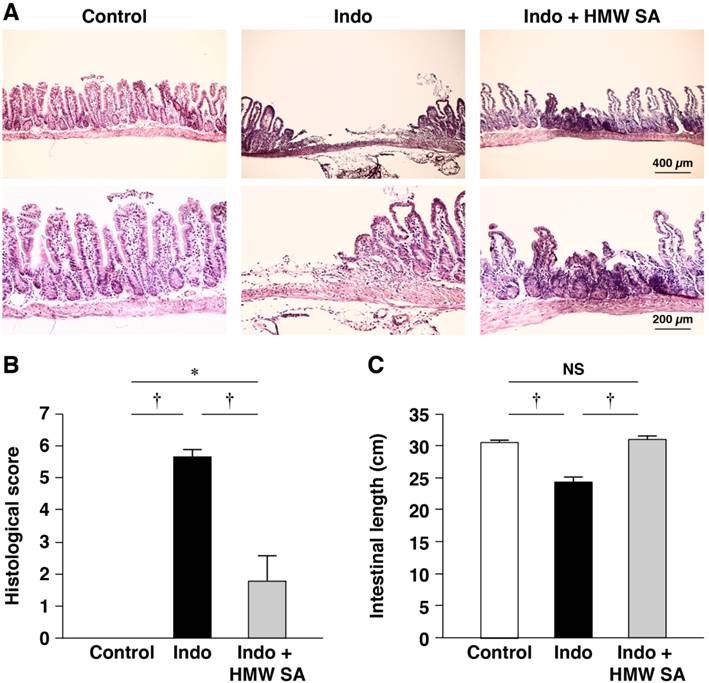
First, we tested the effects of HMW sodium alginate on indomethacin-induced SII in mice. All animals survived under the experimental conditions and body weight between groups was not changed significantly (data not shown). Histology revealed indomethacin to have caused multiple, deep intestinal ulcerations with destruction to the epithelium and infiltration of inflammatory cells within 24 h of administration (Figure 2A). Histological damage scores were increased significantly in indomethacin-treated mice (P<0.01, Figure 2B). Simultaneously, the length of the small intestine in indomethacin-treated mice was significantly shorter than that in control animals (Figure 2C). Pretreatment with HMW sodium alginate before indomethacin administration improved the severity of intestinal lesions significantly (Figure 2A, B) as well as shortening of the length of the small intestine (Figure 2C). CKC is used to measure agreement between the evaluations of two investigators. A negative CKC value suggests that the investigators are rating in the opposite direction, whereas a positive CKC value suggests that the investigators are concordant. The CKC was 0.876, which suggested almost perfect agreement in our analysis. Collectively, these results suggested that HMW sodium alginate reduced the histological severity of indomethacin-induced SII.
Reduction of the histological severity of indomethacin-induced small-intestinal injury in mice by high-molecular-weight sodium alginate (HMW SA). Indomethacin (Indo, 10 mg/kg) was administered to mice that had been given the regular CE-2 or CE-2 premixed with 5% HMW SA for 1 week. Representative low (upper) and high (lower) magnified histological images (A), histological damage scores (B) and the length of the small intestine (C) are shown. †P<0.01, NS: not significant.
Suppression of small-intestinal inflammation by HMW sodium alginate
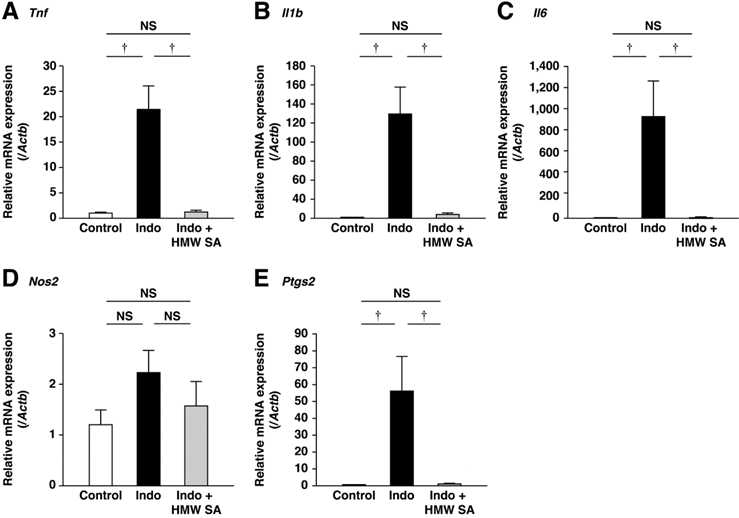
Interactions between luminal bacteria and mucosal immune cells (e.g., neutrophils, macrophages) cause indomethacin-induced intestinal injury. Therefore, we examined if HMW sodium alginate could suppress small-intestinal inflammation. Indomethacin upregulated mRNA expression of inflammatory cytokines such as tumor necrosis factor-α, interleukin-1β and interleukin-6 in the small intestine (Figure 3A-C). HMW sodium alginate ameliorated upregulation of these genes significantly (P<0.01, Figure 3A-C). Similarly, indomethacin upregulated mRNA expression of COX-2, but not iNOS (Figure 3D, E). COX-2 induction was ameliorated significantly by HMW sodium alginate (P<0.01, Figure 3D, E). Thus, HMW sodium alginate suppressed small-intestinal inflammation.
Suppression of small-intestinal inflammation by high-molecular-weight sodium alginate (HMW SA). mRNA expressions of Tnf (A), Il1b (B), Il6 (C), Nos2 (D) and Ptgs2 (E) in the small intestine were analyzed by real-time polymerase chain reaction (PCR). †P<0.01, NS: not significant.
Prevention of indomethacin-induced mucin depletion in the small intestine by HMW sodium alginate
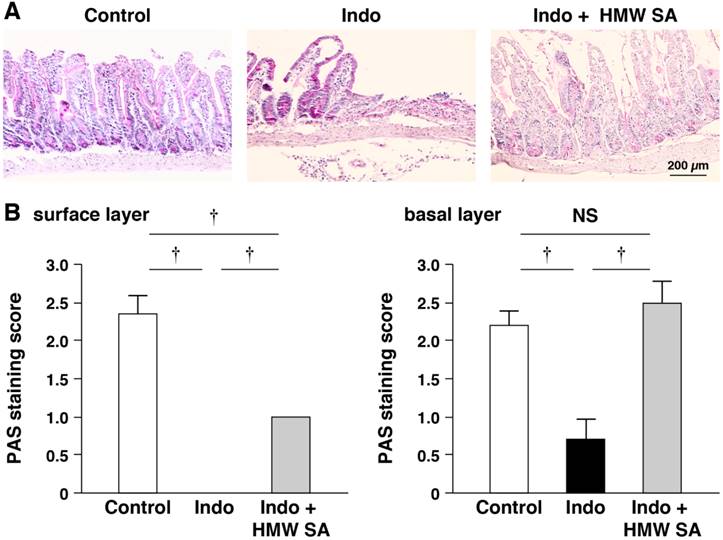
We examined the pattern of PAS staining of goblet cells (major source of mucins). PAS staining showed that indomethacin reduced the number of PAS-stained cells on the surface and in basal layers of small-intestinal ulcers, suggesting depletion of goblet cells (Figure 4A, B). HMW sodium alginate ameliorated the reduction in the number of PAS-stained cells in these layers significantly (P<0.01, Figure 4A, B).
Prevention of indomethacin-induced mucin depletion in the small intestine by high-molecular-weight sodium alginate (HMW SA). Representative periodic acid-Schiff (PAS) staining images (A) and PAS staining scores (B) in both surface and basal (C) layers of small-intestinal ulcers are shown. †P<0.01, NS: not significant.
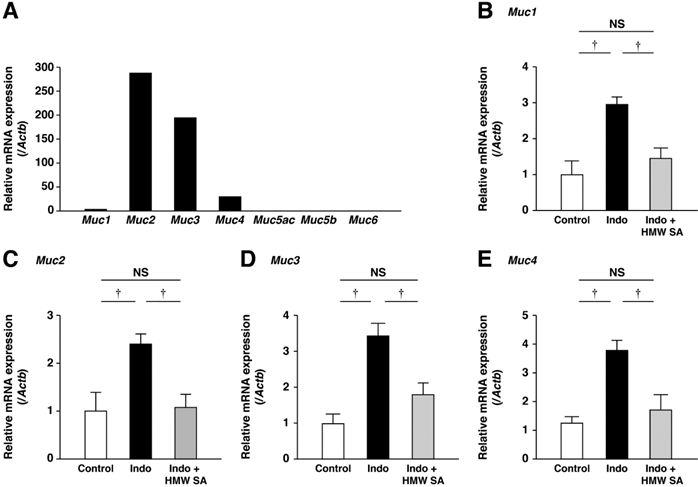
We analyzed mRNA expression of Muc1-Muc6 in untreated control mice. mRNAs of secretory Muc2 and membrane-associated Muc3 and Muc4 were expressed prominently (Figure 5A). Muc1 mRNA was also expressed but mRNAs of Muc5ac, Muc5b, and Muc6 were not. In the small intestine, mRNA expression of Muc1, Muc2, Muc3 and Muc4 was increased, and these increases were decreased significantly by HMW sodium alginate (P<0.01, Figure 5B-D). Thus, HMW sodium alginate prevented indomethacin-induced mucin depletion in the small intestine.
Prevention of the upregulation of mucin genes in the small intestines by high-molecular-weight sodium alginate (HMW SA). mRNA expressions of mucins in the normal small intestine were analyzed using real-time polymerase chain reaction (PCR) (A). mRNA expressions of Muc1 (B), Muc2 (C), Muc3 (D) and Muc4 (E) in the small intestine were analyzed using real-time PCR. †P<0.01, NS: not significant.
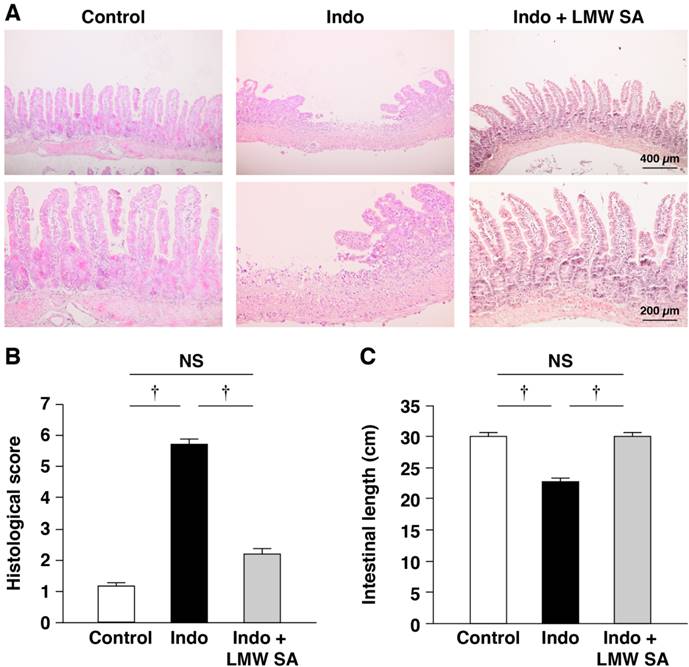
Reduction of the histological severity of indomethacin-induced SII in mice by LMW sodium alginate
Next, we examined if LMW sodium alginate could prevent indomethacin-induced SII in mice. Similar to the preventative effects of HMW sodium alginate, LMW sodium alginate improved the severity of intestinal lesions significantly (P<0.01, Figure 6A, B) as well as shortening the length of the small intestine (Figure 6C). The CKC value was 0.635, which suggested substantial agreement in our analysis. Collectively, these results suggested that LMW sodium alginate reduced the histological severity of indomethacin-induced SII.
Reduction of the histological severity of indomethacin-induced small-intestinal injury in mice by low-molecular-weight sodium alginate (LMW SA). Indomethacin (10 mg/kg) was administered to mice that had been given the regular CE-2 or CE-2 premixed with 5% LMW SA for 1 week. Representative low (upper) and high (lower) magnified histological images (A), histological damage scores (B) and the length of the small intestine (C) are shown. †P<0.01.
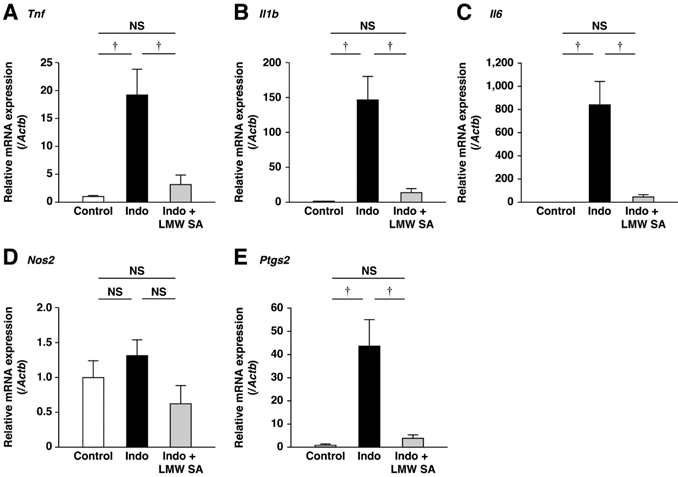
Suppression of small-intestinal inflammation by LMW sodium alginate
Indomethacin-induced upregulation of expression of inflammatory cytokine mRNA was decreased significantly by LMW sodium alginate (P<0.01, Figure 7A-C). Similarly, indomethacin-induced upregulation of expression of inflammatory mediators was ameliorated significantly by LMW sodium alginate (P<0.01, Figure 7D, E). These results suggested that LMW sodium alginate suppressed small-intestinal inflammation.
Suppression of low-molecular-weight sodium alginate (LMW SA) on small-intestinal inflammation by low-molecular-weight sodium alginate (LMW SA). mRNA expressions of Tnf (A), Il1b (B), Il6 (C), Nos2 (D) and Ptgs2 (E) in the small intestine were analyzed using real-time polymerase chain reaction (PCR). †P<0.01, NS: not significant.
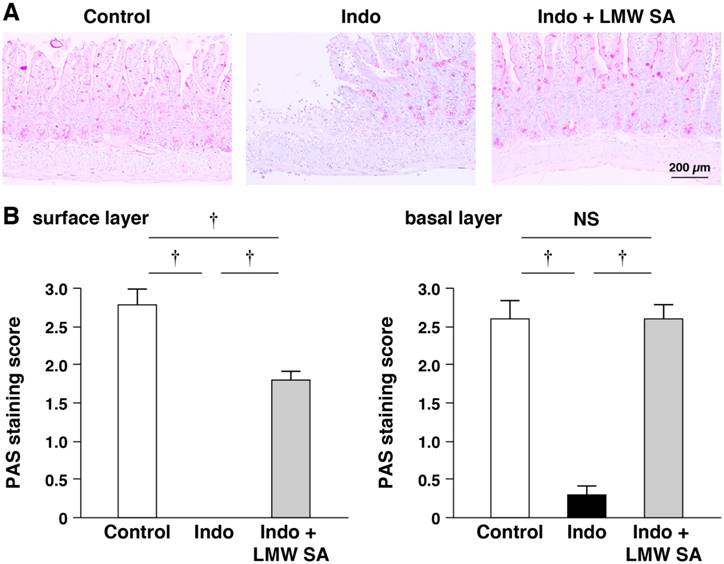
Prevention of indomethacin-induced mucin depletion in the small intestine by LMW sodium alginate
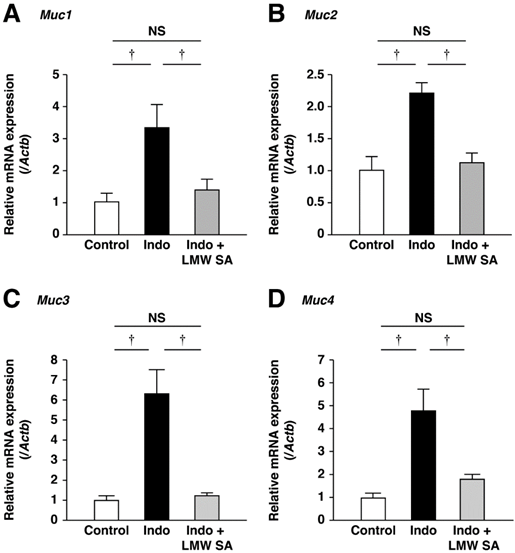
LMW sodium alginate ameliorated reduction in the number of PAS-stained cells on the surface and basal layers of small-intestinal ulcers significantly (P<0.01, Figure 8A, B). Indomethacin-increased mRNA expression of Muc1, Muc2, Muc3 and Muc4 was decreased by LMW sodium alginate (Figure 9A-C). Thus, LMW sodium alginate prevented indomethacin-induced mucin depletion in the small intestine.
Prevention of indomethacin-induced mucin depletion in the small intestine by low-molecular-weight sodium alginate (LMW SA). Representative periodic acid-Schiff (PAS) staining images (A) and PAS staining scores (B) in both surface and basal (C) layers of small-intestinal ulcers are shown. †P<0.01.
Prevention of the upregulation of mucin genes in the small intestine by low-molecular-weight sodium alginate (LMW SA). mRNA expressions of Muc1 (A), Muc2 (B), Muc3 (C) and Muc4 (D) in the small intestine were analyzed using real-time polymerase chain reaction (PCR). †P<0.01, NS: not significant.
Discussion
We showed that pretreatment with HMW sodium alginate ameliorated indomethacin-induced SII in mice. We noted reduction of ≈70% in terms of four histologic factors (width of the ulceration; depth of the lesion; degree of inflammatory infiltration; thrombi), which is similar to the protective action of this drug on gastric ulcers. The etiology of indomethacin-induced SII is more complicated than that of gastric ulcers. Indomethacin inhibits the production of prostaglandin E2, thereby enhancing small-intestinal motility and reducing mucus secretion [10], which allows increasing the intestinal permeability. Intestinal bacteria can invade the mucosa, which triggers consequent inflammation and injury in the small intestine. Sodium alginate, which is not be absorbed in the small intestine, can adhere to mucosal surface of the small intestine owing to its viscosity, which prevents the invasion of intestinal bacteria into the small intestine, thus reducing consequent intestinal inflammation and injury. The viscosity of sodium alginate is likely important for the adhesion to the mucosal surface in small intestine.
Satoh et al reported a correlation between the viscosity and the muco-protective actions of soluble dietary fibers [21]. In this way, sodium alginate could suppress indomethacin-induced SII in mice. In agreement with this hypothesis, HMW sodium alginate has been shown to improve inflammation in the lower GIT in an experimental model of colitis [15, 22] and in methotrexate-induced SII [16]. Here, we also showed that, in addition to HMW sodium alginate, pretreatment with LMW sodium alginate ameliorated indomethacin-induced SII in mice. Protective effects of LMW sodium alginate were comparable with those of HMW sodium alginate even though their viscosities are different. Similar to HMW sodium alginate, these protective effects are probably due to the mucosal-protective action of LMW sodium alginate caused by its adhesion to the mucosal surface and its apophthegmatic action. Thus, our results demonstrate that HMW sodium alginate and LMW sodium alginate have protective effects against indomethacin-induced SII in mice.
We analyzed mucin expression in the small intestine and confirmed mRNA expression of Muc1, Muc2, Muc3, and Muc4, results that are consistent with those of other scholars [23, 24]. In the small intestine, Muc2 is the major component of the secreted mucous barrier and is produced by goblet cells and Paneth cells [23, 24]. Muc5 and Muc6 were secreted by the colon, but not by the small intestine. mRNAs of membrane-associated Muc1, Muc3, and Muc4 were expressed in the small intestine. We also showed that mRNAs of Muc1, Muc2, Muc3, and Muc4 were upregulated in indomethacin-induced SII. These results were consistent with the previous reports showing that mRNAs of Muc1, Muc2, Muc3 and Muc4 were induced by inflammatory cytokines [18, 25, 26]. This phenomenon was probably due to compensation of depletion of mucosal mucins. Consistent with this hypothesis, mRNAs of Muc1, Muc2, Muc3, and Muc4 were ameliorated in indomethacin-treated mice by HMW sodium alginate and LMW sodium alginate, and presumably reflected non-depletion of mucosal mucins.
Scholars have examined the effects of several drugs on indomethacin-induced SII in rats. An opener of KATP channels, diazoxide, ameliorates, whereas a blocker of KATP channels, glibenclamide, enhances injury [27]. Diazoxide exerts its beneficial effects by protecting against mitochondrial damage, reducing intercellular permeability, and relaxing smooth muscle. Rebamipide, an agent that can protect the gastric mucosa, exerts its beneficial effects by inhibiting the genes of matrix metalloproteinases and chemokines [28]. We reported that 18β-glycyrrhetinic acid (an active metabolite of glycyrrhizin that is a major water-soluble constituent of licorice root) and hydroxypropyl γ-cyclodextrin protect against indomethacin-induced SII in mice via an anti-inflammatory mechanism [17]. Thus, several mechanisms may underlie indomethacin-induced SII.
Recently, Yamamoto et al. examined the effects of sodium alginate on indomethacin-induced gastrointestinal mucosal injuries in rats [4]. They administered sodium alginate per os (p.o.) 30 min before and 6 h after indomethacin administration (p.o.) and, 24 h later, assessed lesion formation in the stomach and small intestine. They showed that sodium alginate ameliorated indomethacin-induced mucosal injury in these organs. Sodium alginate ameliorated indomethacin-induced atrophic changes, increases in the number of intestinal bacteria, vascular permeability, oxidative stress, and mucin depletion in the small intestine. In the present study, we provide evidence to support a mechanism by which sodium alginate protects against depletion of mucosal mucins. Therefore, prevention of mucin depletion induced by NSAIDs is a therapeutic strategy for NSAID-induced SII. Mucins exert an intestinal-barrier function to prevent intestinal bacterial translocation [29], which is a major source of pathogenicity arising from NSAID-induced enteritis [30]. Accordingly, protection against depletion of mucosal mucins by sodium alginate may prevent intestinal bacteria infiltration from injury sites in the small intestine. Sodium alginate is already available for human use. Our results suggest that the pre-administration of sodium alginate before administration of NSAIDs is effective for the prevention or reduction of NSAID-induced SII. When pre-administration of sodium alginate is difficult in clinical setting, co-treatment of sodium alginate might be effective for the treatment of NSAID-induced SII. However, it is unknown whether sodium alginate is effective for the prevention or treatment of NSAID-induced SII in humans, so clinical trials to explore the effectiveness of sodium alginate on NSAID-induced SII in humans should be conducted.
Conclusions
Pretreatment with HMW sodium alginate or LMW sodium alginate before indomethacin administration in mice ameliorated mRNA expression of inflammation-related molecules and protected indomethacin-induced mucin depletion in the small intestine. HMW sodium alginate and LMW sodium alginate prevented indomethacin-induced SII. Pretreatment with sodium alginate is effective prophylaxis for NSAID-induced SII.
Acknowledgements
This study was supported in part by a Grant-in-Aid (number 26460212) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (TT). TT was supported financially by KOSEI-KAKENHI (H25-B sou-Kan-en-general-018).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Allison MC, Howatson AG, Torrance CJ. et al. Gastrointestinal Damage Associated with the Use of Nonsteroidal Antiinflammatory Drugs. N Engl J Med. 1992;327:749-54
2. Graham DY, Opekun AR, Willingham FF, Qureshi WA. Visible small-intestinal mucosal injury in chronic NSAID users. Clin Gastroenterol Hepatol. 2005;3:55-9
3. Maiden L, Thjodleifsson B, Theodors A. et al. A quantitative analysis of NSAID-induced small bowel pathology by capsule enteroscopy. Gastroenterology. 2005;128:1172-8
4. Yamamoto A, Itoh T, Nasu R, Nishida R. Sodium alginate ameliorates indomethacin-induced gastrointestinal mucosal injury via inhibiting translocation in rats. World J Gastroenterol. 2014;20:2641-52
5. Higuchi K, Umegaki E, Watanabe T. et al. Present status and strategy of NSAIDs-induced small bowel injury. J Gastroenterol. 2009;44:879-88
6. Wallace JL, Syer S, Denou E. et al. Proton pump inhibitors exacerbate NSAID-induced small intestinal injury by inducing dysbiosis. Gastroenterology. 2011;141:1314-22 22.e1-5
7. Yasuda M, Kawahara R, Hashimura H. et al. Dopamine D(2)-receptor antagonists ameliorate indomethacin-induced small intestinal ulceration in mice by activating alpha7 nicotinic acetylcholine receptors. J Pharmacol Sci. 2011;116:274-82
8. Kawahara R, Yasuda M, Hashimura H. et al. Activation of alpha7 nicotinic acetylcholine receptors ameliorates indomethacin-induced small intestinal ulceration in mice. Eur J Pharmacol. 2011;650:411-7
9. Ikawa Y, Fujino H, Otake S, Murayama T. Indomethacin antagonizes EP(2) prostanoid receptor activation in LS174T human colon cancer cells. Eur J Pharmacol. 2012;680:16-21
10. Kunikata T, Tanaka A, Miyazawa T. et al. 16,16-Dimethyl prostaglandin E2 inhibits indomethacin-induced small intestinal lesions through EP3 and EP4 receptors. Dig Dis Sci. 2002;47:894-904
11. Hatazawa R, Ohno R, Tanigami M. et al. Roles of endogenous prostaglandins and cyclooxygenase isozymes in healing of indomethacin-induced small intestinal lesions in rats. J Pharmacol Exp Ther. 2006;318:691-9
12. Boelsterli UA, Redinbo MR, Saitta KS. Multiple NSAID-induced hits injure the small intestine: underlying mechanisms and novel strategies. Toxicol Sci. 2013;131:654-67
13. Takeuchi K, Tanaka A, Kato S. et al. Roles of COX inhibition in pathogenesis of NSAID-induced small intestinal damage. Clin Chim Acta. 2010;411:459-66
14. Jass JR, Walsh MD. Altered mucin expression in the gastrointestinal tract: a review. J Cell Mol Med. 2001;5:327-51
15. Yamamoto A, Itoh T, Nasu R, Nishida R. Effect of sodium alginate on dextran sulfate sodium- and 2,4,6-trinitrobenzene sulfonic acid-induced experimental colitis in mice. Pharmacology. 2013;92:108-16
16. Yamamoto A, Itoh T, Nasu R. et al. Sodium alginate inhibits methotrexate-induced gastrointestinal mucositis in rats. Biol Pharm Bull. 2013;36:1528-34
17. Ishida T, Miki I, Tanahashi T. et al. Effect of 18beta-glycyrrhetinic acid and hydroxypropyl gammacyclodextrin complex on indomethacin-induced small intestinal injury in mice. Eur J Pharmacol. 2013;714:125-31
18. Imaoka H, Ishihara S, Kazumori H. et al. Exacerbation of indomethacin-induced small intestinal injuries in Reg I-knockout mice. Am J Physiol Gastrointest Liver Physiol. 2010;299:G311-9
19. Linden SK, Florin TH, McGuckin MA. Mucin dynamics in intestinal bacterial infection. PLoS One. 2008;3:e3952
20. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45
21. Satoh H. Role of dietary fiber in formation and prevention of small intestinal ulcers induced by nonsteroidal anti-inflammatory drug. Curr Pharm Des. 2010;16:1209-13
22. Mirshafiey A, Khodadadi A, Rehm BH. et al. Sodium alginate as a novel therapeutic option in experimental colitis. Scand J Immunol. 2005;61:316-21
23. Ho SB, Niehans GA, Lyftogt C. et al. Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res. 1993;53:641-51
24. Ho SB, Shekels LL, Toribara NW. et al. Mucin gene expression in normal, preneoplastic, and neoplastic human gastric epithelium. Cancer Res. 1995;55:2681-90
25. Li X, Wang L, Nunes DP. et al. Pro-inflammatory cytokines up-regulate MUC1 gene expression in oral epithelial cells. J Dent Res. 2003;82:883-7
26. Mejias-Luque R, Linden SK, Garrido M. et al. Inflammation modulates the expression of the intestinal mucins MUC2 and MUC4 in gastric tumors. Oncogene. 2010;29:1753-62
27. Menozzi A, Pozzoli C, Poli E. et al. Diazoxide attenuates indomethacin-induced small intestinal damage in the rat. Eur J Pharmacol. 2011;650:378-83
28. Yamada S, Naito Y, Takagi T. et al. Rebamipide ameliorates indomethacin-induced small intestinal injury in rats via the inhibition of matrix metalloproteinases activity. J Gastroenterol Hepatol. 2012;27:1816-24
29. Frankel W, Zhang W, Singh A. et al. Fiber: effect on bacterial translocation and intestinal mucin content. World J Surg. 1995;19:144-8 discussion 8-9
30. Porras M, Martin MT, Yang PC. et al. Correlation between cyclical epithelial barrier dysfunction and bacterial translocation in the relapses of intestinal inflammation. Inflamm Bowel Dis. 2006;12:843-52
Author contact
![]() Corresponding author: Yoshiyuki Rikitake, MD, PhD. Department of Medical Pharmaceutics, Kobe Pharmaceutical University, 4-19-1 Motoyamakita-machi, Higashinada-ku, Kobe 658-8558, Japan; tel: +81-78-441-7578; fax: +81-78-441-7578; e-mail: rikitakeac.jp.
Corresponding author: Yoshiyuki Rikitake, MD, PhD. Department of Medical Pharmaceutics, Kobe Pharmaceutical University, 4-19-1 Motoyamakita-machi, Higashinada-ku, Kobe 658-8558, Japan; tel: +81-78-441-7578; fax: +81-78-441-7578; e-mail: rikitakeac.jp.