Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2016; 13(3):169-178. doi:10.7150/ijms.13581 This issue Cite
Research Paper
Palmitate-induced Regulation of PPARγ via PGC1α: a Mechanism for Lipid Accumulation in the Liver in Nonalcoholic Fatty Liver Disease
Hitoshi Maruyama ![]() , Soichiro Kiyono, Takayuki Kondo, Tadashi Sekimoto, Osamu Yokosuka
, Soichiro Kiyono, Takayuki Kondo, Tadashi Sekimoto, Osamu Yokosuka
Department of Gastroenterology and Nephrology, Chiba University Graduate School of Medicine, 1-8-1, Inohana, Chuou-ku, Chiba, 260-8670, Japan
Received 2015-8-17; Accepted 2015-11-11; Published 2016-2-11
Abstract
The aim was to examine the effect of free fatty acids on the regulation of PPARγ-PGC1α pathway, and the effect of PPARγ/PGC1α in NAFLD. The mRNA and protein expression of PGC1α and phospho/total PPARγ were examined in Huh7 cells after the palmitate/oleate treatment with/without the transfection with siRNA against PGC1a. The palmitate content, mRNA and protein expression of PGC1α and PPARγ in the liver were examined in the control and NAFLD mice. Palmitate (500 μM), but not oleate, increased protein expression of PGC1α and phospho PPARγ (PGC1α, 1.42-fold, P=0.038; phospho PPARγ, 1.56-fold, P=0.022). The palmitate-induced PPARγ mRNA expression was reduced after the transfection (0.46‑fold), and the protein expressions of PGC1α (0.52-fold, P=0.019) and phospho PPARγ (0.43-fold, P=0.011) were suppressed in siRNA-transfected cells. The palmitate (12325.8 ± 1758.9 μg/g vs. 6245.6 ± 1182.7 μg/g, p=0.002), and mRNA expression of PGC1α (11.0 vs. 5.5, p=0.03) and PPARγ (4.3 vs. 2.2, p=0.0001) in the liver were higher in high-triglyceride liver mice (>15.2 mg/g) than in low-triglyceride liver mice (<15.2 mg/g). The protein expressions of both PGC1α and PPARγ were higher in the NAFLD group than in the controls (PGC1α, 1.41-fold, P=0.035; PPARγ, 1.39-fold, P=0.042), and were higher in the high-triglyceride liver group (PGC1α, 1.52-fold, p=0.03; PPARγ, 1.22-fold, p=0.05) than in the low-triglyceride liver group. In conclusion, palmitate appear to up-regulate PPARγ via PGC1α in Huh7 cells, and both PGC1α and PPARγ are up-regulated in the NAFLD mice liver, suggesting an effect on lipid metabolism leading to intrahepatic triglyceride accumulation.
Keywords: Palmitate, peroxisome proliferator-activated receptor γ, peroxisome proliferator-activated receptor coactivator 1 α, triglyceride, liver, nonalcoholic fatty liver disease
Introduction
Nonalcoholic fatty liver disease (NAFLD) is increasing worldwide as one of the leading causes of chronic liver diseases [1-3]. The condition comprises nonalcoholic fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH), both with hepatic steatosis; the latter is distinguished from the former by the presence of cytological ballooning and inflammation on histology [4, 5]. NAFLD is closely associated with obesity, diabetes, hyperlipidemia, physical inactivity, and a high‑fat diet [6-8]. Although the mechanism is still unclear, free fatty acids (FFA) may play a critical role in the development of NAFLD [9-13].
The PPARs (peroxisome proliferator-activated receptors) belong to the nuclear receptor superfamily. There are 3 subtypes in the PPAR family, PPARα, PPARδ/β, and PPARγ, and tissue distribution varies depending on the subtype: PPARα is found mainly in liver, heart, and kidney; PPARγ mainly in adipose tissue; and PPARδ is ubiquitously-distributed [14-16]. They function as transcription factors which control the expression of genes involved in fat and glucose metabolism, and cellular proliferation and differentiation. They act by binding to the promoter of the target gene after forming a heterodimer with the retinoid X receptor. Previous studies have shown a close relationship between PPAR and clinical presentations such as diabetes, obesity, and inflammation [17]. Various biological functions regulated by PPARγ may account for the principal mechanisms for type 2 diabetes [18] and arteriosclerosis [19-21].
The peroxisome proliferator-activated receptor coactivator 1 (PGC1) comprises a family of transcriptional coactivators, including PGC1α, PGC1β, and the PGC related coactivator (PRC) [22]. PGC1α shows an interaction with transcriptional factors like PPARα, PPARγ, estrogen-related receptor, liver X receptor, and hepatocyte nuclear factor-4α. In addition, PGC1α functions as a regulator of mitochondrial metabolism [23]. It regulates energy, glucose and fat metabolism, and is recognized as an important therapeutic target for diabetes and obesity.
Based on these backgrounds, we hypothesized that PGC1α and PPARγ may have an interactive effect on the pathogenesis of NAFLD. The study investigated the expression of PGC1α and PPARγ in FFA-treated culture cells, and measured the content of palmitate and expression of PGC1α and PPARγ in NAFLD mice with respect to the triglyceride content. The aim of this study was to examine the in vivo and in vitro effect of fatty acid via PGC1α and PPARγ in the pathogenesis of NAFLD.
Materials and Methods
Cell culture
A human hepatoma cell line (Huh7) was used in the study. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-activated fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin.
FFA treatment
Two FFAs were used in the study, palmitate and oleate; they were purchased from Sigma Chemical Company (St. Louis, MO). The cells (500,000 cells/well) seeded in the 6-well plates were incubated with each of FFAs mixed with 5% bovine serum albumin at final concentrations of 100-1000 μM.
Cell transfection
Huh7 cells were seeded on 6-well plates, and transfection with siRNA against PGC1α (sc-38884) was performed according to the manufacturer's protocol (Santa Cruz Biotechnology, Inc., Dallas, TX). The scrambled sequence that does not lead to the specific degradation of any known cellular mRNA (sc-37007) was used as a control. Huh7 cells were incubated for 6 h with the transfection reagent, and normal growth medium containing serum and antibiotics was added for overnight incubation. Then, the medium was replaced with normal growth medium and the cells were used for FFA-treatment procedures 1 day later.
Animal model for NAFLD
The study used six- to ten-week-old male STAMTM mice, which were purchased from Stelic Institute & Co. (Charles River Laboratories, Japan Inc.) and sacrificed to obtain liver tissue. The mouse model was established by the following protocol supported by the similar procedure in the literature [24]; 2-day-old male pups were injected with streptozotocin (200 μg per mouse) and started on a high-fat diet (HFD-32) from the age of 4 weeks. The animals develop steatosis to steatohepatitis from 6 to 8 weeks of age, and fibrosis from 9 to 12 weeks of age, showing various grades of NAFLD. The study also used control mice under control diet. The mice were euthanized by the inhalation of methoxyflurane to take the blood and liver sample. Animal care and study protocols were approved by the Animal Care Committee of Chiba University.
Real-time quantitative reverse transcription polymerase chain reaction
Total RNA in the cell or tissue was extracted using TRIzol reagent according to the manufacturer's protocol (Invitrogen, Carlsbad, CA). Single-strand cDNAs were synthesized from 2 μg total RNA in a 20 μL reaction (SuperScript® VILOTM, cDNA Synthesis Kit, Invitrogen). Polymerase chain reactions (PCR) were performed using cDNA, SYBR green (Platinum® SYBR® Green qPCR SuperMix-UDG with ROX, Invitrogen) and primers for PGC1α, PPARγ, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, endogenous control), purchased from Takara Bio (Tokyo, Japan; Table 1, 2). Reactions were run in triplicate and data were calculated as the change in cycle threshold (ΔCT) for the target gene relative to the ΔCT for GAPDH.
Primers for quantitative polymerase chain reaction (human)
| Gene | Forward/reverse | Sequence 5'-3' |
|---|---|---|
| PGC1α | Forward | GGAGACGTGACCACTGACAATGA |
| Reverse | TGTTGGCTGGTGCCAGTAAGAG | |
| PPARγ | Forward | TTGAAAGAAGCCAACACTAAACCAC |
| Reverse | AATGGCATCTCTGTGTCAACCAT | |
| GAPDH | Forward | GCACCGTCAAGGCTGAGAAC |
| Reverse | TGGTGAAGACGCCAGTGGA |
Protein extraction and western blot analysis
Cell lysates and liver samples after homogenization were centrifuged at 12,000 g for 15 min and proteins in the supernatants were used for western blotting to detect PGC1α, phospho PPARγ (S112) and PPARγ.
Proteins were separated using 4%-12% NuPAGE® Novex Bis-Tris Mini Gels (Invitrogen) and were transferred to a nitrocellulose membrane for 1.5 h at 40 V using a western blot apparatus (Invitrogen). After overnight incubation with primary antibody, the membranes were washed and then incubated with horseradish peroxidase-conjugated secondary antibodies. Proteins were detected with an enhancement using SuperSignal chemiluminescence reagent (Pierce Biotechnology, Inc., Rockford, IL) and the density was quantified using an LAS-4000UV (Fuji Film, Tokyo, Japan) and Adobe Photoshop (CS4; Adobe Systems, San Jose, CA). Primary antibodies were purchased as follows: phospho PPARγ and PPARγ from Abcam® (Tokyo, Japan) and PGC1α from Santa Cruz Biotechnology, Inc. (Dallas, TX). A secondary antibody and β-actin were purchased from Cell Signaling (Beverly, MA).
Primers for quantitative polymerase chain reaction (mouse)
| Gene | Forward/reverse | Sequence 5'-3' |
|---|---|---|
| PGC1α | Forward | ACACAACGCGGACAGAATTGAG |
| Reverse | TCACAGGTGTAACGGTAGGTGATG | |
| PPARγ | Forward | GGAGCCTAAGTTTGAGTTTGCTGTG |
| Reverse | TGCAGCAGGTTGTCTTGGATG | |
| GAPDH | Forward | TGTGTCCGTCGTGGATCTGA |
| Reverse | TTGCTGTTGAAGTCGCAGGAG |
Quantification of lipid accumulation
Lipid accumulation in the cultured cells was quantitatively assessed using Steatosis Colorimertic Assay kit (Cayman Chemical Company, MI). After overnight incubation of 5,000 cells/well in 96 well plates, the cells were transfected with siRNA against PGC1α or scrambled RNA, both followed by 24-h palmitate treatment (500 μM). The cells were stained according to the manufacture's protocol, and lipid accumulation was determined by the absorbance at 490nm. The lipid accumulation was expressed as a ratio of FFA-treated cells to control (untreated cells).
Lipid analysis in the mouse liver tissue
Lipids were extracted from liver tissue (approximately 100 mg per mouse) according to Folch's method with chloroform/methanol [25]. Triglyceride and total cholesterol were quantified using Cholestest® (Sekisui Medical Corp. Tokyo, Japan). Total fatty acid content (free and esterified, μg/g) in the liver tissue was measured by gas chromatography (GC profiles) with the samples prepared by chloroform and methanol using GC-2010 Plus (Shimadzu, Kyoto, Japan).
Statistical analysis
Data are presented by mean ± standard deviation, or range. Continuous variables were compared by the Student's t-test or Fisher's Protected Least Significant Difference test. P-values less than 0.05 were considered statistically significant in all analyses. Statistical analysis was performed using the Dr. SPSS software package (version 11.0J for Windows; SPSS Inc., Chicago, Illinois, USA).
Results
Changes in mRNA expression of PGC1α after palmitate treatment in Huh7 cells
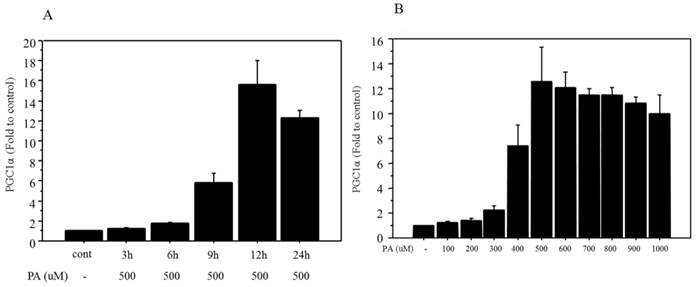
Firstly, a 500 μM dose of palmitate was used according to the literatures [26,27]. The mRNA expression of PGC1α showed incubation time-related changes and maximum expression (15.7‑fold change vs. control) was observed after 12 h of treatment (Figure 1A). Next, changes in mRNA expression of PGC1α were examined after 12 h of treatment with different doses of palmitate, ranging from 100 to 1000 μM. The mRNA expression of PGC1α showed dose-related changes and maximum expression (12.5‑fold change vs. control) was observed at a dose of 500 μM palmitate (Figure 1B).
Changes in mRNA expression of PGC1α after palmitate treatment. A. The mRNA of PGC1α showed incubation time-related changes following 500 μM of palmitate treatment, and maximum expression (15.7‑fold change vs. control) was observed with 12 h of treatment. B. The mRNA of PGC1α showed dose-related changes (100 to 1000 μM), and maximum expression (12.5‑fold change vs. control) was observed at a dose of 500 μM after 12 h of palmitate treatment. Data are expressed as mean ± standard deviation. Four independent experiments were performed to generate the results. PA, palmitate
Changes in mRNA expression of PGC1α after oleate treatment in Huh7 cells
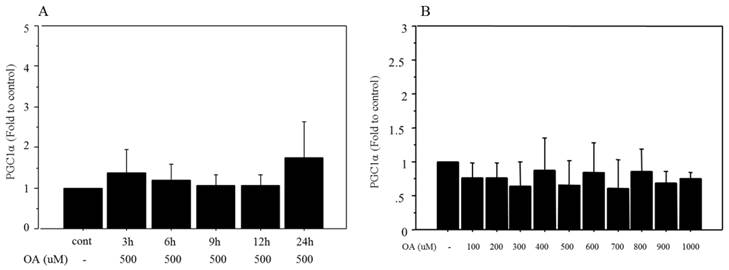
Time-related changes in mRNA expression of PGC1α were also examined after treatment with 500 μM oleate. There were no significant changes in the expression after oleate treatment (Figure 2A). Similarly, mRNA expression of PGC1α showed no significant changes after 12 h of treatment with different doses of oleate, ranging from 100 to 1000 μM (Figure 2B).
Changes in mRNA expression of PGC1α after oleate treatment. A. There were no significant changes in PGC1α following 500 μM of oleate treatment B. PGC1α showed no significant changes following 12 h of treatment with different doses of oleate, ranging from 100 to 1000 μM. Data are expressed as mean ± standard deviation. Four independent experiments were performed to generate the results. OA, oleate
Changes in mRNA expression of PPARγ after FFA treatment
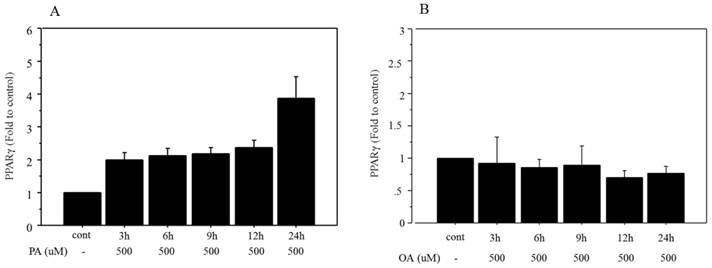
Time-related changes in mRNA expression of PPARγ were examined after treatment with 500 μM palmitate and the maximum increase (3.92‑fold change vs. control, Figure 3A) was seen after 24 h of incubation. However, the mRNA expression of PPARγ did not show any significant change after treatment with 500 μM oleate over an incubation time ranging from 3 to 24 h (Figure 3B).
Changes in mRNA expression of PPARγ after FFA treatment. A. Time-related changes of mRNA expression of PPARγ were examined after incubation with 500 μM palmitate, and maximum increase (3.92‑fold change vs. control) was seen after 24 h of treatment. PA, palmitate. B. The mRNA expression of PPARγ did not show any significant changes after treatment with 500 μM of oleate, over incubation times ranging from 3 to 24 h. OA, oleate. Data are expressed as mean ± standard deviation. Four independent experiments were performed to generate the results.
Regulation of mRNA expression of PPARγ by PGC1α
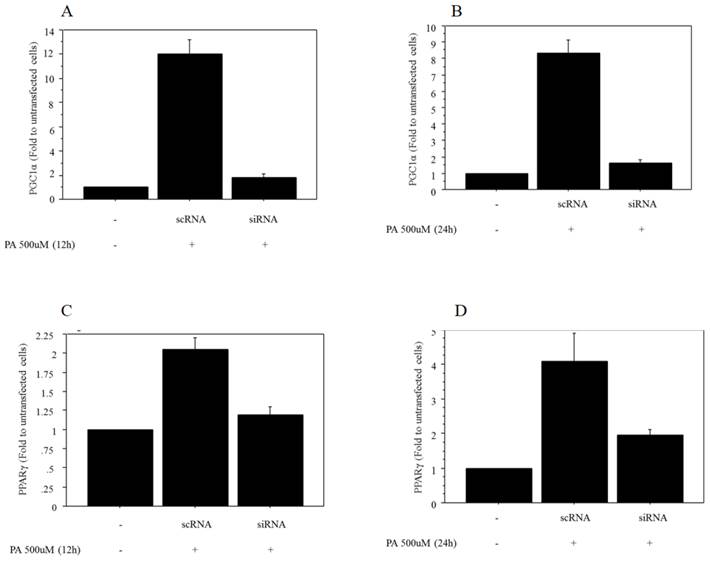
The mRNA expression of PGC1α after 500 μM of palmitate treatment was significantly decreased in Huh7 cells transfected with siRNA against PGC1α (0.17‑fold vs. control after 12 h of treatment, 0.23‑fold vs. control after 24 h of treatment) (Figure 4A, 4B). The mRNA expression of PPARγ after 500 μM of palmitate treatment showed a significant decrease in Huh7 cells after transfection with siRNA against PGC1α (0.51‑fold vs. control after 12 h of treatment, 0.46‑fold vs. control after 24 h of treatment) (Figure 4C, 4D).
Regulation of mRNA expression of PPARγ by PGC1α. A. The mRNA expression of PGC1α after 12 h of treatment with 500 μM palmitate showed a significant decrease in Huh7 cells transfected with siRNA against PGC1α (0.17‑fold vs. control transfected with scRNA). B. The mRNA expression of PGC1α after 24 h of treatment with 500 μM palmitate showed a significant decrease in Huh7 cells transfected with siRNA against PGC1α (0.23‑fold vs. control transfected with scRNA). C. The mRNA expression of PPARγ after 12 h of treatment with 500 μM palmitate showed a significant decrease in Huh7 cells after transfection with siRNA against PGC1α (0.51‑fold vs. control transfected with scRNA). D. The mRNA expression of PPARγ after 24 h of treatment with 500 μM palmitate showed a significant decrease in Huh7 cells after transfection with siRNA against PGC1α (0.46‑fold vs. control transfected with scRNA). Data are expressed as mean ± standard deviation. Six independent experiments were performed to generate the results. PA, palmitate.
Lipid accumulation in Huh7 cells
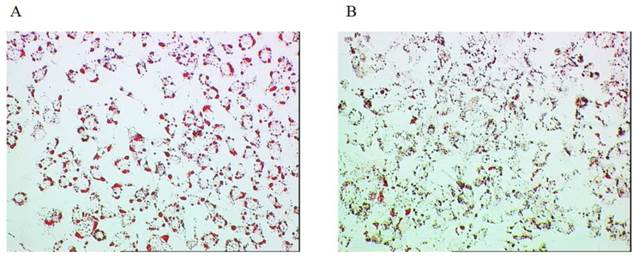
Lipid accumulation was significantly lower in the cells transfected with siRNA (1.34 ± 0.21) than those transfected with scramble RNA (1.68 ± 0.25, p=0.031, n=7) both followed by 24-h palmitate treatment (500 μM) (Figure 5A, 5B).
Lipid accumulation in the cultured cell. A. Huh7 cells transfected with scramble RNA followed by 24-h palmitate treatment (500 μM). B. Huh7 cells transfected with siRNA followed by 24-h palmitate treatment (500 μM).
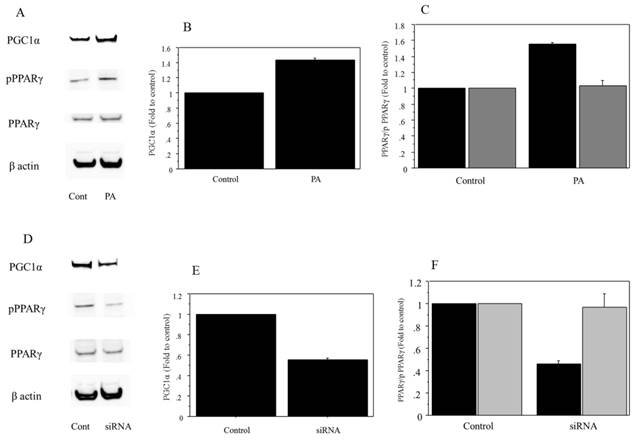
Protein analysis
Analysis of protein extracts showed that 500 μM of palmitate treatment induced a significant increase in the expression of PGC1α and phospho PPARγ (PGC1α, 1.42-fold vs. control, P=0.038; phospho PPARγ, 1.56-fold vs. control, P=0.022) (Figure 6A, B, C). The expression was suppressed in Huh7 cells transfected with siRNA against PGC1α (PGC1α, 0.52-fold vs. scramble RNA as control, P=0.019; phospho PPARγ, 0.43-fold vs. control, P=0.011) (Figure 6D, E, F). There was no significant change in the expression of total PPARγ in the palmitate-treated Huh7 cells.
Protein analysis. A. The 500μM of palmitate treatment induced an increase in the expression of PGC1α and phospho PPARγ. There was no expression change in the total PPARγ in the palmitate-treated Huh7 cells. Control, untreated cells; PA, palmitate. The gels shown are representative of four independent experiments. B. The 500μM of palmitate treatment induced a significant increase in the expression of PGC1α (1.42-fold vs. control, P=0.038). Control, untreated cells; PA, palmitate. The data in the graphs are expressed as the ratio of the target protein to β-actin (n=4). C. The 500μM of palmitate treatment induced a significant increase in phospho PPARγ (phospho PPARγ, 1.56-fold vs. control, P=0.022). There was no significant change in the expression of total PPARγ in the palmitate-treated Huh7 cells. Control, untreated cells; PA, palmitate; Black for phospho PPARγ, grey for total PPARγ. The data in the graphs are expressed as the ratio of the target protein (phospho PPARγ/ total PPARγ) to total PPARγ or β-actin (n=4). D. The protein expression in PGC1α, phospho PPARγ and total PPARγ was compared between control and cells transfected with siRNA against PGC1α, both treated with palmitate. The expression in PGC1α and phospho PPARγ was suppressed in the Huh7 cells transfected with siRNA against PGC1α. There was no expression change in the total PPARγ. Control, cells transfected with scRNA. The gels shown are representative of four independent experiments. E. The protein expression in PGC1α was compared quantitatively between control and cells transfected with siRNA against PGC1α, both treated with palmitate. The expression of PGC1α was significantly suppressed in Huh7 cells transfected with siRNA against PGC1α (PGC1α, 0.52-fold vs. control, P=0.019). Control, cells transfected with scRNA. The data in the graphs are expressed as the ratio of the target protein to β-actin (n=4). F. The protein expression in phospho PPARγ and total PPARγ was compared quantitatively between control and cells transfected with siRNA, both treated with palmitate. The expression of phospho PPARγ was significantly suppressed in the Huh7 cells transfected with siRNA against PGC1α (phospho PPARγ, 0.43-fold vs. control, P=0.011). There was no significant change in the expression of total PPARγ. Control, cells transfected with scRNA; Black for phospho PPARγ, grey for total PPARγ. The data in the graphs are expressed as the ratio of the target protein (phospho PPARγ/ total PPARγ) to total PPARγ or β-actin (n=4).
Lipid analysis in mouse liver tissue
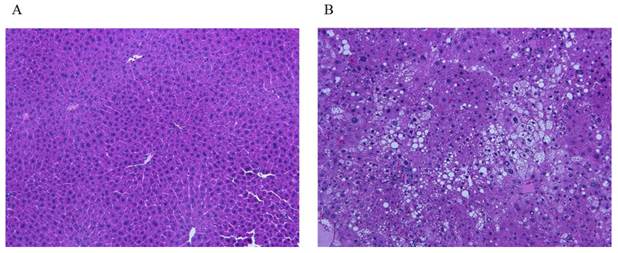
The study examined 16 mice: 4 control mice and 12 mice for NAFLD model (Figure 7; A control, B steatohepatitis model). Blood test showed significant difference in total cholesterol and FFA between control (71 ± 9.9 mg/dl, 979 ± 178 μEq/L) and NAFLD model (134 ± 31 mg/dl, 2463 ± 777μEq/L, Table 3).
Liver tissue images of mice. A. Control. B. The image shows steatosis, lobular inflammation, and ballooning, indicating a presence of steatohepatitis. The images show typical findings of controls (n=4) and NAFLD mice (n=12).
Blood test in the mice.
| Control | NAFLD | P value | |
|---|---|---|---|
| Total cholesterol (mg/dl) | 71±9.9 | 134±31 | 0.04 |
| Triglyceride (mg/dl) | 48±6.4 | 270±448 | 0.53 |
| Free fatty acid (μEq/L) | 979±178 | 2463±777 | 0.044 |
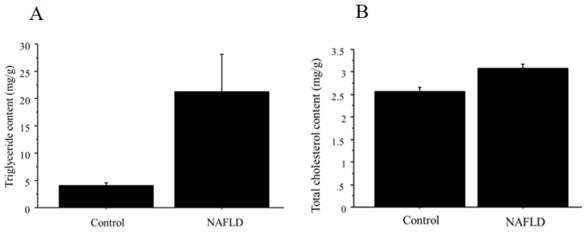
The content of triglyceride and total cholesterol in the liver is summarized in Figure 8 (A, triglyceride; B, total cholesterol), showing significant difference between control (n=4; triglyceride; 4.0 ± 1.4 mg/g, total cholesterol, 2.6 ± 0.17 mg/g) and NAFLD mice (n=12; triglyceride; 21.3 ± 18 mg/g, p=0.026; total cholesterol; 3.1 ± 0.22 mg/g, p=0.0032).
A. The content of triglyceride in the liver tissue. Triglyceride showed significant difference between control (n=4; 4.0 ± 1.4 mg/g) and NAFLD mice (n=12; 21.3 ± 18 mg/g, p=0.026). B. The content of total cholesterol in the liver tissue. Total cholesterol showed significant difference between control (n=4; 2.6 ± 0.17 mg/g) and NAFLD mice (n=12; 3.1 ± 0.22 mg/g, p=0.0032).
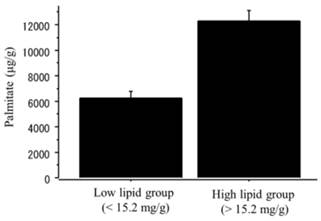
The content of palmitate was significantly higher in the NAFLD group than in the controls (NAFLD, 14750.3 ± 5268.6 μg/g; controls, 5678.5 ± 678.6 μg/g, p=0.01). When the NAFLD mice was divided into the two groups according to the median value of triglyceride content (15.2 mg/g), the content of palmitate was significantly greater in the high-triglyceride liver group (n=6; > 15.2 mg/g; palmitate 12325.8 ± 1758.9 μg/g) than in the low-triglyceride liver group (n=6; <15.2 mg/g; palmitate 6245.6 ± 1182.7 μg/g, p=0.002) (Figure 9).
The content of palmitate in the liver tissue. The NAFLD mouse group was divided into two groups according to the median value of triglyceride content (15.2 mg/g). The palmitate was significantly greater in the high-triglyceride liver group (n=6; > 15.2 mg/g; palmitate 12325.8 ± 1758.9 μg/g) than in the low-triglyceride liver group (n=6; <15.2 mg/g; palmitate 6245.6 ± 1182.7 μg/g, p=0.002).
The mRNA and protein expression of PPARγ and PGC1α in mouse liver tissue
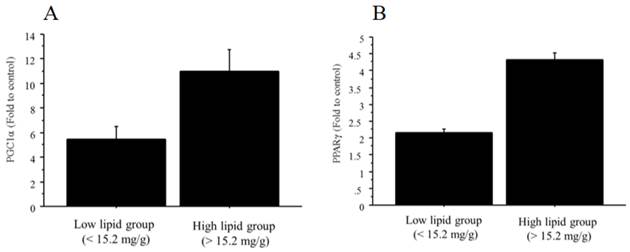
The mRNA expressions of both PGC1α and PPARγ were significantly higher in the NAFLD group than in the controls (PGC1α, 9.36‑fold change vs. control; PPARγ, 4.12‑fold change vs. control). In addition, the mRNA expressions of PGC1α (11.0 ± 3.6 vs. 5.5 ± 1.9, fold to control, p=0.03) and PPARγ (4.3 ± 0.4 vs. 2.2 ± 0.2, fold to control, p=0.0001) were significantly greater in the high-triglyceride liver group (n=6; > 15.2 mg/g) than in the low-triglyceride liver group (n=6; <15.2 mg/g) (Figure 10A, B). There was no significant relationship between total cholesterol content and PPARγ/PGC1α.
The mRNA expression of PGC1α and PPARγ in liver tissue. A. The NAFLD mouse group was divided into two groups according to the median value of triglyceride content (15.2 mg/g). The mRNA expression of PGC1α was significantly greater in the high-triglyceride liver group (n=6; > 15.2 mg/g; 11.0 ± 3.6, fold to control) than in the low-triglyceride liver group (n=6; <15.2 mg/g; 5.5 ± 1.9, fold to control, p=0.03). B. The NAFLD mouse group was divided into two groups according to the median value of triglyceride content (15.2 mg/g). The mRNA expression of PPARγ was significantly greater in the high-triglyceride liver group (n=6; > 15.2 mg/g; 4.3 ± 0.4, fold to control) than in the low-triglyceride liver group (n=6; <15.2 mg/g; 2.2 ± 0.2, fold to control, p=0.0001).
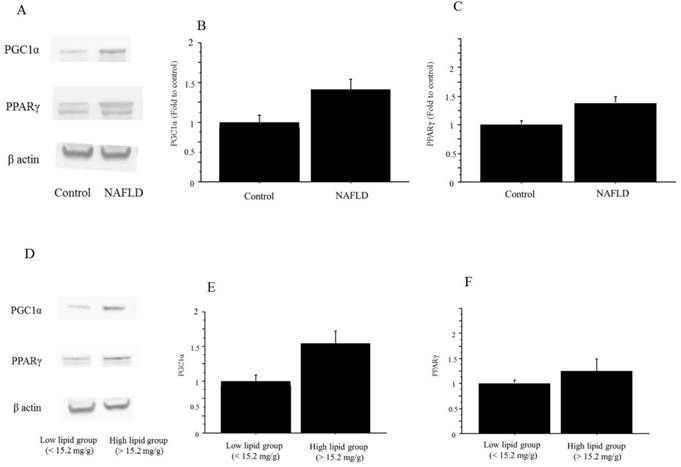
The protein expressions of both PGC1α and PPARγ were significantly higher in the NAFLD group than in the controls (PGC1α, 1.41-fold to control, P=0.035; PPARγ, 1.39-fold vs. control, P=0.042) (Figure 11). Similarly, the protein expressions in both PGC1α and PPARγ were higher in the high-triglyceride liver group (n=6; PGC1α, 1.52-fold, p=0.03; PPARγ, 1.22-fold, p=0.05) than in the low-triglyceride liver group (n=6), the difference in the expression in the former was significant but that in the latter remained marginal (Figure 11).
The protein expression of PGC1α and PPARγ in liver tissue. A. The protein expressions in both PGC1α and PPARγ were higher in the NAFLD mice than in the control mice. The gels shown are representative of 6 independent experiments. B. The protein expression of PGC1α was significantly higher in the NAFLD group (n=12) than in the controls (n=4; PGC1α, 1.41-fold to control, P=0.035). The data in the graphs are expressed as the ratio of the target protein to β-actin. C. The protein expression of both PPARγ was significantly higher in the NAFLD group (n=12) than in the controls (n=4; PPARγ, 1.39-fold vs. control, P=0.042). The data in the graphs are expressed as the ratio of the target protein to β-actin. D. The protein expressions in PGC1α and PPARγ were higher in the high-triglyceride liver group than in the low-triglyceride liver group in the NAFLD mice. The gels shown are representative of 6 independent experiments. E. The protein expression of PGC1α was significantly higher in the high-triglyceride liver group (n=6; 1.52-fold, P=0.03) than in the low-triglyceride liver group (n=6) in the NAFLD mice. The data in the graphs are expressed as the ratio of the target protein to β-actin. F. The protein expression of PPARγ was significantly higher in the high-triglyceride liver group (n=6; 1.22-fold vs. control, P=0.05) than in the low-triglyceride liver group (n=6) in the NAFLD mice. The data in the graphs are expressed as the ratio of the target protein to β-actin.
Discussion
Despite of the continuous effort on the research, the mechanisms for NAFLD/NASH remain unclear [3, 13]. The present study focused on the biological function of FFA in the liver cell line and demonstrated the significant effect of palmitate on the intrahepatic triglyceride accumulation via PGC1a-PPAR pathway. The linkage of the content between palmitate and triglyceride was also proved in the animal model. In the various possible pathogenesis, FFA may play a major role for developing NAFLD which are associated with an impaired hepatic metabolism and triglyceride accumulation in the liver [10-12, 28].
PGC1α acts as a regulator of energy metabolism, such as mitochondrial biogenesis and respiration, adaptive thermogenesis, and gluconeogenesis [22, 29]. One of the major functions of PGC1α is a detoxification of reactive oxygen species which are generated during mitochondrial respiration, resulting in the increase of mitochondrial functions [30-32]. Exercise, low temperatures, and fasting are physiological conditions that stimulate PGC1α expression [22]. The present study demonstrated the additional function of PGC1α, a potential mediator of the palmitate effect of lipid metabolism. At this point, some studies have focused on the FFA-related changes of PGC1α expression. One study showed that unsaturated FFA increased the mRNA expression of PGC1α by 2- to 3-fold in human skeletal muscle cells, though saturated FFA did not affect the mRNA expression of PGC1α [33]. Another two studies reported the palmitate-induced reduction of mRNA expression of PGC1α; one showed that exposure of C2C12 skeletal muscle cells to 0.75 mmol/l palmitate, but not to oleate, reduced PGC-1α mRNA levels (66%; p<0.001), through a mechanism involving MAPK-extracellular signal-regulated kinase (ERK) and NF-κB activation [34]. Palmitate-induced reduction of PGC1α and β expression by 38% (p=0.01) and 53% (p=0.006), respectively, via p38 MAPK-dependent transcriptional pathways in C2C12 myotubes has also been reported [35]. These data may contradict the results in our study performed in human liver cells, suggesting a different function of PGC1α in the liver in response to palmitate treatment.
The present study demonstrated the increased level of PPARγ in both cultured cell model and mice model. Previous studies have also shown increased mRNA expression of PPAR in the obesity-related liver, PPARγ in ob/ob mice [36], and PPARα and PPARγ in murine models of obesity [37]. The authors of the former study reported that lean mice expressed only low levels of PPARγ1 and barely detectable amounts of PPARγ2. However, obese animals showed a marked increase of PPARγ2, with low levels of PPARγ1. Therefore, they speculated that the peroxisome proliferator-like effects of rosiglitazone in obese mice may be due to activation of PPARγ2. A recent human study also reported that mRNA expression of PPARγ was significantly higher in obese patients (n=22, NAFLD) compared with controls. Furthermore, PPARγ expression in the liver showed positive associations with sterol regulatory element binding protein 1c mRNA levels, serum insulin levels, and homeostasis model assessment-insulin resistance, and negative correlations with total adiponectin [38]. These data strongly suggest the role of PPARγ in the development of NAFLD, supporting the results in our study.
As for the biochemical function of PPAR, the current study stresses the effect on the fat accumulation. There are some studies focusing on this issue; one study showed that PPARγ-deficient liver in ob/ob mice was smaller and had a dramatically decreased triglyceride content compared with equivalent mice lacking the AlbCre transgene (ob/ob-PPARγ(fl/fl)AlbCre-) [39]. The mRNA levels of the hepatic lipogenic genes, fatty acid synthase, acetyl-CoA carboxylase, and stearoyl-CoA desaturase-1 were reduced and the levels of serum triglyceride and FFA were significantly higher in ob/ob-PPARγ(fl/fl)AlbCre+ mice than in the control mice. Another study reported similar findings; mice without liver PPARγ, but with adipose tissue, developed relative fat intolerance, increased adiposity, hyperlipidemia, and insulin resistance [40]. Therefore, the authors concluded that liver PPARγ regulates triglyceride homeostasis, contributing to hepatic steatosis, but protects other tissues from triglyceride accumulation and insulin resistance.
In contrast, a previous study reported the no significant effect of palmitate on the regulation of PPARγ, being inconsistent with our data [41]. Although the exact reason is undetermined, it might be explained by the difference in the experimental conditions, different cell line, different concentration of FFA and bovine serum albumin. At the same time, their study suggested the different influence on the lipid accumulation between palmitate and oleate, which were also detected in our study as the oleate showed no effect on the PGC1α-PPARγ pathway. Nonetheless, the dose of palmitate used in our study may be relatively higher than the actual human environment, that is the major limitation of the study, a substantial in vivo effect of FFA need to be validated in the additional studies.
In summary, the current study has shown that palmitate, but not oleate, up-regulates PPARγ via PGC1α in Huh7 cells. Furthermore, both PGC1α and PPARγ are up-regulated and palmitate content was increased in the liver in the NAFLD mouse model showing a positive relationship with triglyceride content, suggesting a certain effect on lipid metabolism leading to intrahepatic triglyceride accumulation. The findings may enhance a better understanding of the pathogenesis of developing NAFLD/NASH and indicate future therapeutic targets for the disease.
Abbreviations
NAFLD: Nonalcoholic fatty liver disease
NAFL: Nonalcoholic fatty liver
NASH: Nonalcoholic steatohepatitis
FFA: Free fatty acids
PPAR: Peroxisome proliferator-activated receptor,
PGC1: Peroxisome proliferator-activated receptor coactivator 1
PRC: PGC related coactivator
DMEM: Dulbecco's Modified Eagle's Medium
PCR: Polymerase chain reactions
GAPDH: Glyceraldehyde-3-phosphate dehydrogenase
Competing Interests
The authors have declared that no competing interest exists.
References
1. Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387-1395
2. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ; American Gastroenterological Association; American Association for the Study of Liver Diseases. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the american gastroenterological association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592-1609
3. Yki-Järvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014;2:901-910
4. Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434-438
5. Cortez-Pinto H, de Moura MC, Day CP. Non-alcoholic steatohepatitis: from cell biology to clinical practice. J Hepatol. 2006;44:197-208
6. Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD single topic conference. Hepatology. 2003;37:1202-1219
7. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(Suppl):S99-S112
8. Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alfa expression via a lysosomal pathway. Hepatology. 2004;40:185-194
9. Carter-Kent C, Zein NN, Feldstein AE. Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: implications for treatment. Am J Gastroenterol. 2008;103:1036-1042
10. Feldstein AE. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin Liver Dis. 2010;30:391-401
11. Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142:711-725
12. Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10:627-636
13. Ganz M, Szabo G. Immune and inflammatory pathways in NASH. Hepatol Int. 2013;7:S771-781
14. Zhu Y, Alvares K, Huang Q, Rao MS, Reddy JK. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J Biol Chem. 1993;268:26817-26820
15. Braissant O, Foufellie F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPARα, -b and -g in the adult rat. Endocrinology. 1996;137:354-366
16. Jain S, Pulikuri S, Zhu Y, Qi C, Kanwar YS, Yeldandi AV, Rao MS, Reddy JK. Differential expression of the peroxisome proliferator-activated receptor g (PPAR g) and its coactivators steroid receptor coactivator-1 and PPAR-binding protein PBP in the brown fat, urinary bladder, colon and breast of the mouse. Am J Pathol. 1998;153:349-354
17. Bortolini M, Wright MB, Bopst M, Balas B. Examining the safety of PPAR onists - current trends and future prospects. Expert Opin Drug Saf. 2013;12:65-79
18. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557-566
19. Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferatorsactivated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523-531
20. Chen Z, Ishibashi S, Perrey S, Osuga J, Gotoda T, Kitamine T, Tamura Y, Okazaki H, Yahagi N, Iizuka Y, Shionoiri F, Ohashi K, Harada K, Shimano H, Nagai R, Yamada N. Troglitazone inhibits atherosclerosis in apolipoprotein E-knockout mice: pleiotropic effects on CD36 expression and HDL. Arterioscler Thromb Vasc Biol. 2001;21:372-377
21. Collins AR, Meehan WP, Kintscher U, Jackson S, Wakino S, Noh G, Palinski W, Hsueh WA, Law RE. Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:365-371
22. Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728-735
23. Austin S, St-Pierre J. PGC1α and mitochondrial metabolism-emerging concepts and lerevance in aging and neurodegenerative disorders. J Cell Sci. 2012;125:4963-4971
24. Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, Arumugam S, Watanabe K, Ichida T, Asakura H, Yoneyama H. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol. 2013;46:141-152
25. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497-509
26. Wei Y, Wang D, Gentile CL, Pagliassotti MJ. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol Cell Biochem. 2009;331:31-40
27. Maruyama H, Takahashi M, Sekimoto T, Shimada T, Yokosuka O. Linoleate appears to protect against palmitate-induced inflammation in Huh7 cells. Lipids Health Dis. 2014;1:78
28. Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081-1090
29. Huss JM, Kopp RP, Kelly DP. Peroxisome Proliferator-activated Receptor Coactivator-1α(PGC-1α) Coactivates the Cardiac-enriched Nuclear Receptors Estrogen-related Receptor-α and γ. J Biol Chem. 2002;277:40265-46274
30. St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, Spiegelman BM. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278:26597-26603
31. Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562-573
32. St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Ja¨ger S, Handschin C, Zheng K, Lin J, Yang W Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397-408
33. Staiger H, Staiger K, Haas C, Weisser M, Machicao F, Haring HU. Fatty acid-induced differential regulation of the genes encoding peroxisome proliferator-activated receptor-γ coactivator-1α and -1β in human skeletal muscle cells that have been differentiated in vitro. Diabetologia. 2005;48:2115-2118
34. Coll T, Jove M, Rodriguez-Calvo R, Eyre E, Palomer X, Sanchez RM, Merlos M, Laguna JC, Vazquez-Carrera M. Palmitate-Mediated Downregulation of Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α in Skeletal Muscle Cells Involves MEK1/2 and Nuclear Factor-κB Activation. Diabetes. 2006;55:2779-2787
35. Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D, Faucette R, Barry K, Bianco AC, Patti ME. Peroxisome Proliferator Activator Receptor γCoactivator-1 expression is reduced in obesity. Potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:15439-15450
36. Edvardsson U, Bergström M, Alexandersson M, Bamberg K, Ljung B, Dahllöf B. Rosiglitazone (BRL49653), a PPAR g-selective agonist, causes peroxisome proliferator-like liver effects in obese mice. J Lipid Res. 1999;40:1177-1184
37. Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C, Feingold KR. Up-Regulation of Peroxisome Proliferator-Activated Receptors (PPAR-a) and PPAR-g Messenger Ribonucleic Acid Expression in the Liver in Murine Obesity: Troglitazone Induces Expression of PPAR-g-Responsive Adipose Tissue-Specific Genes in the Liver of Obese Diabetic Mice. Endocrinology. 2000;141:4021-4031
38. Pettinelli P, Videla LA. Up-Regulation of PPAR-γ mRNA Eexpression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab. 2011;96:1424-1430
39. Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B Jr, Reitman ML, Gonzalez FJ. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111:737-747
40. Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ, Reitman ML. Liver Peroxisome Proliferator-activated Receptor γContributes to Hepatic Steatosis, Triglyceride Clearance, and Regulation of Body Fat Mass. J Biol Chem. 2003;278:34268-34276
41. Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni S, Lonardo A, Carulli N, Loria P. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroenterol Hepatol. 2009;24:830-840
Author contact
![]() Corresponding author: Hitoshi Maruyama. TEL: 81-43-2262083, FAX: 81-43-2262088, E-MAIL: maru-cibac.jp
Corresponding author: Hitoshi Maruyama. TEL: 81-43-2262083, FAX: 81-43-2262088, E-MAIL: maru-cibac.jp