Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(11):891-904. doi:10.7150/ijms.13187 This issue Cite
Research Paper
Fimasartan, a Novel Angiotensin-Receptor Blocker, Protects against Renal Inflammation and Fibrosis in Mice with Unilateral Ureteral Obstruction: the Possible Role of Nrf2
Soojeong Kim1, Sung Jun Kim2, Hye Eun Yoon2, Sungjin Chung1, Bum Soon Choi1, Cheol Whee Park1, Seok Joon Shin2 ![]()
1. Division of of Nephrology, Department of Internal Medicine, College of Medicine, The Catholic University of Korea
2. Division of of Nephrology, Department of Internal Medicine, Incheon St. Mary's Hospital, College of Medicine, The Catholic University of Korea
Received 2015-7-8; Accepted 2015-9-20; Published 2015-10-21
Abstract
Objectives: A newly developed angiotensin II receptor blocker, fimasartan, is effective in lowering blood pressure through its action on the renin-angiotensin system. Renal interstitial fibrosis, believed to be due to oxidative injury, is an end-stage process in the progression of chronic kidney disease. Nuclear factor erythroid 2-related factor 2 (Nrf2) is known to regulate cellular oxidative stress and induce expression of antioxidant genes. In this study we investigated the role of Nrf2 in fimasartan-mediated antioxidant effects in mice with renal fibrosis induced by unilateral ureteral obstruction (UUO).
Materials and Methods: UUO was induced surgically in mice, followed by either no treatment with fimasartan or the intraperitoneal administration of fimasartan (3 mg/kg/day). On day 7, we evaluated the changes in the renin-angiotensin system (RAS) and the expression of Nrf2 and its downstream antioxidant genes, as well as renal inflammation, apoptosis, and fibrosis in the obstructed kidneys. The effect of fimasartan on the Nrf2 pathway was also investigated in HK-2 cells stimulated by tumor necrosis factor-α.
Results: The mice with surgically induced UUO showed increased renal inflammation and fibrosis as evidenced by histopathologic findings and total collagen content in the kidney. These effects were attenuated in the obstructed kidneys of the fimasartan-treated mice. Fimasartan treatment inhibited RAS activation and the expression of Nox1, Nox2, and Nox4. In contrast, fimasartan upregulated the renal expression of Nrf2 and its downstream signaling molecules (such as NQO1; HO-1; GSTa2 and GSTm3). Furthermore, it increased the expression of antioxidant enzymes, including CuSOD, MnSOD, and catalase. The fimasartan-treated mice had significantly less apoptosis on TUNEL staining, with decreased levels of pro-apoptotic protein and increased levels of anti-apoptotic protein. In the HK-2 cells, fimasartan treatment inhibited RAS activation, decreased expression of mitogen-activated protein kinases (MAPKs), and upregulated the Nrf2 pathway.
Conclusions: These results suggest that fimasartan has beneficial effects in reducing renal oxidative stress, inflammation, and fibrosis. Possible mechanisms to explain these effects are inhibition of RAS and MAPKs and upregulation of Nrf2 signaling, with subsequent induction of antioxidant pathways.
Keywords: fimasartan, nuclear factor erythroid-2-related factor 2, renin-angiotensin system, oxidative stress, MAPKs
1. Introduction
Renal tubulointerstitial fibrosis is a common pathologic condition that leads to end-stage renal disease. Several studies have demonstrated that oxidative stress is an important etiologic factor in renal fibrosis. Patients with chronic kidney disease (CKD) have evidence of accumulated oxidative stress caused by interactions between reactive oxygen species (ROS) and proteins, carbohydrates, and lipids [1]. Oxidative stress is known to be strongly associated with increased cardiovascular mortality in patients with CKD and those on dialysis [2]. ROS increase lipid peroxidation and hydrogen peroxides and can injure DNA or protein function, which causes renal tubulointerstitial damage [3]. ROS is also involved in renal fibrosis by regulating inflammatory cell infiltration and extracellular matrix accumulation in the renal interstitium in human and experimental animal models. Therefore, it has been suggested that antioxidant treatment might ameliorate renal fibrosis and thus the progression of CKD [4, 5].
Nuclear factor erythroid 2-related factor 2 (Nrf2) is known to be critical in inducing antioxidant enzymes. It binds to the antioxidant response element (ARE), which is located in the promoter region of genes encoding antioxidant and phase II detoxifying enzymes such as glutathione peroxidase, NAD(P)H:quinone oxidoreductase 1 (NQO1), catalase, heme oxygenase-1 (HO-1), thioredoxin reductase, and the glutamate-cysteine ligase catalytic subunit [6]. Under normal circumstances, Nrf2 is known to be localized in the cytoplasm as an inactive complex form through binding to its repressor molecule, Kelch-like ECH-associated protein1 (Keap1) [7]. Under conditions such as oxidative or endoplasmic reticulum stress, the Keap1-Nrf2 complex is dissociated, and cytoplasmic Nrf2 subsequently translocates to the nucleus and binds to the ARE of genes encoding antioxidant enzymes [8]. The Nrf2-mediated modulation of cellular antioxidant and anti-inflammatory effects plays an important role in protecting against oxidative stress [9].
In the kidney, activation of the renin-angiotensin system (RAS) induces hypertension, recruits proinflammatory or profibrotic cells, and causes the accumulation of extracellular matrix components, resulting in renal damage and CKD progression [10]. Thus, RAS blockade has not only antihypertensive effects but also antioxidative, anti-inflammatory, and antifibrotic effects. Blockade of the RAS with an angiotensin II (ATII) receptor blocker (ARB) is highly effective in alleviating the progression of kidney disease in both humans and experimental animals [11].
Fimasartan, a newly developed ARB, was approved in 2010 for the treatment of essential hypertension in Korea (Boryung Pharm. Co. Ltd, Seoul, Korea). Its molecular formula is C27H31N7OS, [(2-n-butyl-5-dimethylamino-thiocarbonylmethyl-6-methyl-3-[[2′-(1H-tetrazol-5-yl) biphenyl-4-yl] methyl] pyrimidin-4(3H)-one), indicating the bioisosteric replacement of the imidazole part of losartan with pyrimidin-4(3H)-one [12]. Although previous studies of fimasartan have described its safety profile and antihypertensive and antiatherosclerotic effects in human and animal experiments [13], its salutary effect in renal disease has rarely been examined. In this study, we formulate the hypothesis that fimasartan ameliorates oxidative stress, inflammation, and fibrosis in the kidneys of mice with unilateral ureteral obstruction and that upregulation of Nrf2-dependent antioxidative signaling may be involved in this process.
2. Materials and methods
2.1 Experimental animals
For this experiment, we obtained 9-week-old male C57BL/6 mice weighing 23 to 28 g (DooYeol Biotech, Inc., Seoul, Korea). The animals were housed in a controlled-temperature environment with a 12-hour light/dark cycle for one week prior to initiation of the experiments. Mice underwent either sham or UUO operations, as described previously [14]. Fimasartan potassium trihydrate, which was donated by Boryung Pharmaceutical Company (Seoul, Korea), was dissolved in distilled water and administered intraperitoneally (3 mg/kg/day) from the day of UUO surgery and was continued for 7 days after the operation. On day 7, the mice were divided equally into three experimental groups, as follows: sham-operated mice (n=8), UUO-operated control mice day 7 (UUO7) (n=8), and UUO-operated fimasartan-treated mice day 7 (UUO7-FM) (n=8). All the mice were sacrificed. Both the sham and obstructed kidneys were harvested and then processed for the experiments. All the experimental procedures were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of our institute.
2.2 Histologic examination
All the obstructed kidneys were cleared by means of perfusion with phosphate buffered saline (PBS) and were fixed in 10% formalin. Paraffin-embedded kidney tissues were used for histologic analyses. Masson trichrome staining was used to assess the severity of tubulointerstitial fibrosis. More than 15 randomly selected fields from the corticomedullary junction area were evaluated, and images were obtained using a light microscope (Zeiss LSM 510, Carl Zeiss, Jena, Germany). Fibrotic areas were quantified in selected fields with the use of MetaMorph imaging software (Molecular Devices LLC, Downingtown, PA, USA). The severity of tubulointerstitial fibrosis was expressed as the ratio of the fibrotic area to the total selected field.
To determine the extent of inflammatory cell infiltration, we performed immunohistochemical testing for F4/80 and α-smooth muscle antin (α-SMA); to assess the degree of apoptosis, we employed the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, as previously described [15]. Briefly, after deparaffinization and hydration, the sections were incubated in 0.5% Triton X-100-PBS solution and washed with PBS. Normal donkey serum was used to block any nonspecific antibody-binding reactions. The sections were incubated overnight in a humidified chamber at 4°C with primary antibody for F4/80 and α-SMA (all from Abcam, Cambridge, MA, USA). Peroxidase-conjugated anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) was used as a secondary antibody. The sections were incubated with a mixture of 0.05% 3,3′-diaminobenzidine containing 0.01% H2O2 for color reactions. After counterstaining with hematoxylin, the sections were analyzed under light microscopy. Twenty high-power fields that included the renal corticomedullary junction were randomly selected from each section. F4/80-positive cells were counted and proportional areas of α-SMA staining were quantified using MetaMorph imaging software.
The TUNEL assay was performed on paraffin sections fixed with 4% paraformaldehyde according to the manufacturer's instructions (Millipore, Billerica, MA, USA) to identify apoptotic cells. TUNEL-positive cells were counted in cortical tubule cells in 20 fields per slide. All the slides were analyzed in a blinded manner.
2.3 Total collagen assay
The total collagen content of the kidney tissue was measured by acid hydrolysis of the kidney tissue, as described previously [15]. Briefly, each kidney sample was hydrolyzed in 6N HCl for 18 hours at 110°C and then dried at 75°C. Each sample was solubilized in citric acid collagen buffer (0.23 mol/L of citric acid, 0.88 mol/L of sodium acetate trihydrate, 0.85 mol/L of sodium hydroxide, and 1.2% acetic acid) and filtered through a 0.45-μm centrifugal filter unit (Ultrafree-MC, Millipore, Billerica, MA, USA). For the oxidation reaction, 100 μL of chloramine-T solution (1.4% chloramine-T and 10% n-propanol in citric acid buffer) was added to each sample, and 100 μL of Ehrlich's reagent (15% 4-dimethylamino-benzaldehyde, 62% n-propanol, and 18% perchloric acid) was added to start the color reaction. A spectrophotometric assay at 550 nm was performed for measuring the amount of hydroxyproline, and total collagen content in the kidney tissue was calculated on the assumption that collagen contains 12.7% hydroxyproline by sample weight.
2.4 In vitro experiment using HK-2 cells and cell death assessment
Human renal proximal tubule epithelial cells (HK-2 cells) purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA) were cultured in keratinocyte-serum-free media supplemented with recombinant epidermal growth factor and bovine pituitary extract. Cells were split or refreshed with complete media every 3 or 4 days, and cells with passage number 7 or 8 were used for these experiments.
Cell viability was measured by means of a cell proliferation and cytotoxicity assay using Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD, USA). HK-2 cells were seeded at a density of 1×104 cells per well in 96-well plates, and after 24 hours, fimasartan was added to reach different final concentrations. Following incubation for another 24 hours, CCK-8 was employed and the optical density of each well was measured at 450 nm. Cell viability was expressed as relative cell survival, which indicates the percentage of optical density of HK-2 cells treated with fimasartan versus that of the vehicle-treated cells. The concentrations of fimasartan used for cell treatment were 62.5 and 125 µM, which showed a relative cell survival of above 90%. Cells were plated onto 60-mm dishes and were made quiescent in growth factor-free medium for 24 hours. Cells were then treated with tumor necrosis factor-α (TNF-α) (5 ng/mL) either with or without fimasartan.
2.5 Immunofluorescence analysis of HK-2 cells
HK-2 cells were analyzed using the indirect immunofluorescence method. To determine the presence of ATII, the HK-2 cells were grown on coverslips and fixed in 4% (v/v) paraformaldehyde in PBS for 20 minutes at room temperature. Cells were blocked in 2.5% normal horse serum (Vector Laboratories Inc, Burlingame, CA, USA) for 40 minutes at room temperature in a humidified chamber. The cells were then incubated in PBS containing a mixture of 1:50 primary antibody (Santa Cruz Biotechnology) overnight at 4°C in a humidified chamber. Cells were washed in PBS and incubated in goat anti-rabbit secondary antibody for 1 hour at room temperature in a humidified chamber. Cells were washed with PBS and incubated in tryptic soy agar (TSA) (1:50) (PerkinElmer Inc, Waltham, MA, USA) for 5 minutes at room temperature, followed by nuclear staining with 4',6-diamidino-2-phenylindole (DAPI). The immunofluorescence signal was visualized with the use of fluorescent microscopy and was photographed with the use of LSM 700 laser scanning confocal microscopy (Zeiss, Jena, Germany).
2.6 Semiquantitative immunoblotting
The proteins of kidney tissues or HK-2 cells were extracted using the PRO-PREP Protein Extraction Kit (Intron Biotechnology, Inc., Seongnam, Gyeonggi-do, Korea), and nuclear protein extracts were also prepared with the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Rockford, IL, USA) according to the manufacturer's instructions. The protein concentration was determined by means of a Bradford assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
For immunodetection, the blot was incubated overnight at 4°C with the primary antibodies against the following proteins: ATII type 1 receptor (AT1R), Nox1, and Nox4 (all from Santa Cruz Biotechnology); Nox2 (BD Biosciences, San Jose, CA, USA); Keap1 and Nrf2 (all from Santa Cruz Biotechnology); Lamin B1 (Cell Signaling Technology, Beverly, MA, USA); β-actin (Sigma-Aldrich, St. Louis, MO, USA); NQO-1 (Santa Cruz Biotechnology); HO-1 (BD Biosciences); CuSOD, MnSOD, and catalase (all from Abcam); Bax and Bcl-2 (all from Santa Cruz Biotechnology); Bim (Cell Signaling Technology); and mitogen-activated protein kinases (MAPKs), including phospho-JNK, phospho-p38 MAPK, and phospho-ERK1/2 (all from Cell Signaling Technology). The secondary antibodies were goat anti-rabbit IgG with horseradish peroxidase (Cell Signaling Technology) for Keap1, Nrf2, Lamin B1, HO-1, Nox1, Nox2, Nox4, CuSOD, catalase, Bax, Bcl-2, and Bim; horse anti-mouse IgG with horseradish peroxidase (Cell Signaling Technology) for MnSOD and β-actin; and rabbit anti-goat IgG with horseradish peroxidase (Sigma-Aldrich) for NQO1. Protein bands were detected using a chemiluminescence imaging system (Fusion SL4-3500, Vilber Lourmat, Marne-la-Vallée, France), and band densities were determined by Quantity One software (Bio-Rad).
2.7 RNA isolation and reverse transcriptase and quantitative real-time polymerase chain reactions
Total RNA was extracted from kidney tissues or HK-2 cells with the use of TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. A reverse transcriptase reaction was carried out for the synthesis of cDNA, and quantitative real-time polymerase chain reaction (PCR) assays were performed using SYBR Premix (Takara Bio Inc., Otsu, Shiga, Japan). Primer sequences for each mouse gene are listed in Table 1. All polymerase chain reactions were duplicated for each sample.
Sequences of primers used for quantitative polymerase chain reaction analysis of mouse genes
| Gene | Forward (5'-3') | Reverse (5'-3') |
|---|---|---|
| NQO1 | TATCCTTCCGAGTCATCTCTAGCA | TCTGCAGCTTCCAGCTTCTTG |
| HO-1 | ATCGTGCTCGCATGAACACT | CCAACACTGCTATTACATGGC |
| GSTa2 | CGTCCACCTGCTGGAACTTC | GCCTTCAGCAGAGGGAAAGG |
| GSTm3 | GCTCTTACCACGTGCAGCTT | GGCTGGGAAGAGGAAATGGA |
| GPx1 | GATGAACGATCTGCAGAAGC | CAAAGTTCCAGGCAATGTCG |
| GPx2 | TCATGACCGATCCCAAGCTCAT | ATGGCAACTTTAAGGAGGCGCT |
| β-actin | AGTGTGACGTTGACATCCGTA | GCCAGAGCAGTAATCTCCTTCT |
2.8 Statistical analysis
Data are expressed in means ± SE. Statistical differences among groups were calculated using analysis of variance (ANOVA), followed by Bonferroni correction. P-values of < 0.05 were considered to be statistically significant.
3. Results
3.1 Effects of fimasartan in mice with UUO
3.1.1 Effects of fimasartan on systolic blood pressure
Before sham or UUO surgery, the average systolic blood pressure (SBP) was similar among the three groups of mice, and this did not change throughout the entire experimental period (Table 2). The intraperitoneal administration of fimasartan at a dose of 3 mg/kg/day did not significantly lower the average SBP of mice in the UUO group treated with fimasartan (P > 0.05).
Systolic blood pressure (mm Hg)
| Group | Day 0 | Day 7 |
|---|---|---|
| Sham | 103.3±3.9 | 106.7±4.5 |
| UUO7 | 103.5±5.0 | 104.7±13.7 |
| UUO7-FM | 106.0±6.8 | 103.4±16.7 |
Values are means±SE. UUO, unilateral ureteral obstruction; FM, fimasartan.
3.1.2 Effects of fimasartan on renal morphologic changes and markers of fibrosis
On Masson trichrome staining, the extent of extracellular matrix deposition within the renal tubulointerstitial area was found to be increased after UUO. Fimasartan treatment significantly ameliorated UUO-induced tubulointerstitial fibrosis in the obstructed kidneys obtained from the UUO7-FM mice (UUO7 vs. UUO7-FM, P = 0.018) (Fig. 1A and 1B).
Effect of fimasartan (FM) on renal morphologic changes, inflammatory cell infiltration, and renal fibrosis in unilateral ureteral obstruction (UUO). (A) Representative photographs assessing tubulointerstitial fibrosis (Masson trichrome, ×200), immunohistochemical staining for the infiltration of F4/80-positive cells (×400) and for α-SMA (×400). (B) Semiquantitative analysis of fibrotic areas. (C) Number of F4/80-positive cells. (D) Semiquantitative analysis of α-SMA-positive area. (E) The amount of hydroxyproline in renal tissue. UUO7 = UUO-control day 7; UUO7-FM = UUO-fimasartan day 7. *P < 0.05 vs. sham and UUO7. †P < 0.05 vs. UUO7. Values are expressed as means ± SE.
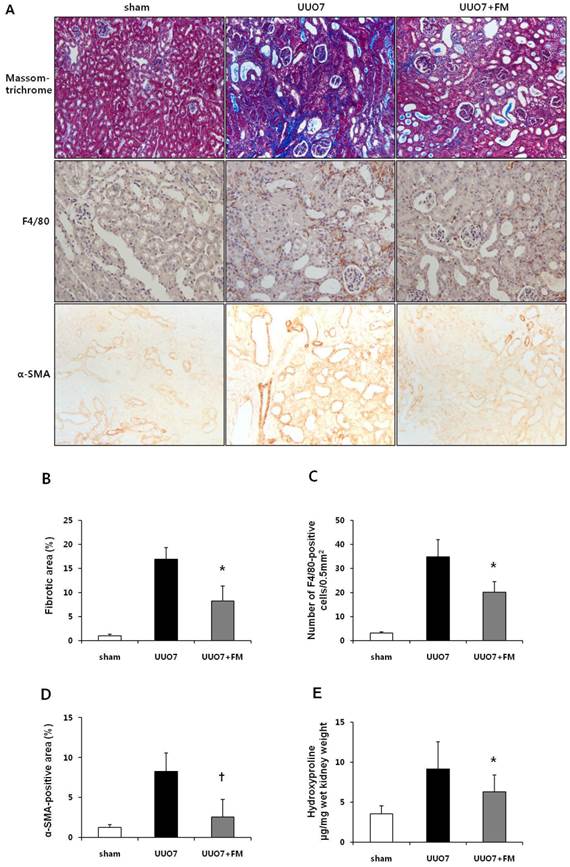
Staining for F4/80 revealed the degree of interstitial macrophage infiltration (Fig. 1A). The number of F4/80-positive cells markedly increased in the obstructed kidneys of the UUO mice. Fimasartan also decreased UUO-induced infiltration of F4/80-positive cells (UUO7 vs. UUO7-FM, 34.9±7.1 vs. 20.2±4.3 cells/0.5 mm2, P = 0.026) (Fig. 1C).
Immunohistochemical testing for α-SMA, a marker of myofibroblasts, showed a pattern in keeping with the severity of tubulointerstitial fibrosis (Fig. 1A). The α-SMA-positive areas that had increased after UUO were significantly decreased in the obstructed kidneys of the fimasartan-treated UUO mice (UUO7 vs. UUO7-FM, P = 0.039) (Fig. 1D).
In the obstructed kidneys, the amount of hydroxyproline, which reflects total collagen content in renal tissue, was found to be increased after UUO. Fimasartan prominently decreased these amounts in the obstructed kidneys from UUO-FM mice (UUO7 vs. UUO7-FM, 9.13±3.40 vs. 6.27±2.12 hydroxyproline µg/mg wet kidney weight, P = 0.042) (Fig. 1E).
3.1.3 Renal expression of AT1R and Nox signaling
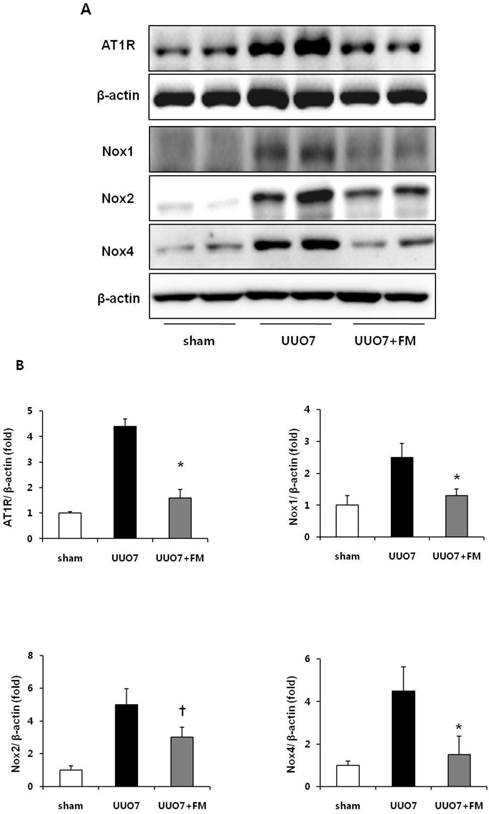
The protein levels of AT1R, Nox1, Nox2, and Nox4 were significantly increased in group UUO7 compared with those in the sham group, and these changes were significantly attenuated in the UUO7-FM group (UUO7 vs. UUO7-FM: AT1R, P = 0.012; Nox1, P = 0.043; Nox2, P = 0.039; Nox4, P = 0.044) (Fig. 2A and 2 B).
Effect of fimasartan (FM) on the expression of AT1R and Nox in unilateral ureteral obstruction (UUO). (A) Representative photographs of immunoblots of AT1R, Nox1, Nox2, and Nox4. (B) The immunofold of the expression of AT1R, Nox1, Nox2, and Nox4. UUO7 = UUO-control day 7; UUO7-FM = UUO-fimasartan day 7. *P < 0.05 vs. UUO7. †P < 0.05 vs. sham and UUO7. Values are expressed as means ± SE.
3.1.4 Keap1 and Nrf2 signaling
The expression of intrarenal Keap1 and of Nrf2 proteins was evaluated using immunoblot analysis (Fig. 3A). Intrarenal Keap1 expression was increased in the UUO group compared with the sham mice, and treatment with fimasartan did not significantly change the level of Keap1 expression compared with the UUO7 group (UUO7 vs. UUO7-FM, P > 0.05).
Effect of fimasartan (FM) on the expression of Keap1 and Nrf2 in unilateral ureteral obstruction (UUO). (A) Representative immunoblots of Keap1, total Nrf2, and cytoplasmic and nuclear Nrf2. (B) The immunofold of the expression of Keap1 and the ratio of the expression of nuclear Nrf2 to that of cytoplasmic Nrf2. UUO7 = UUO-control day 7; UUO7-FM = UUO-fimasartan day 7. *P < 0.05 vs. sham and UUO7. Values are expressed as means ± SE.
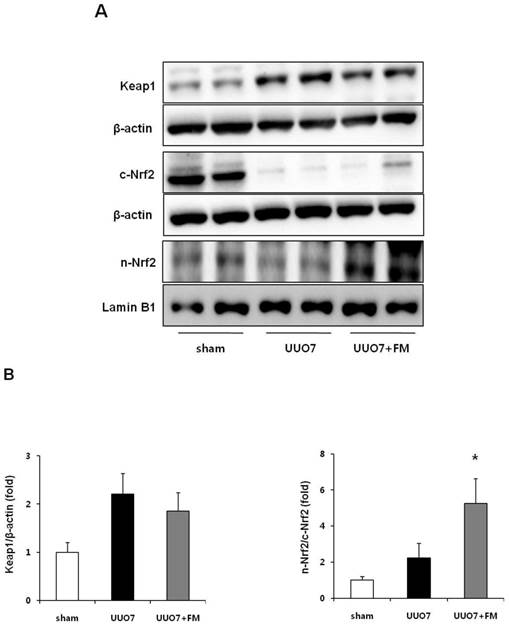
UUO did not decrease the expression of nuclear Nrf2, whereas the expression of cytoplasmic Nrf2 was significantly decreased after UUO. Interestingly, fimasartan failed to increase the expression of cytoplasmic Nrf2; however, it markedly enhanced the expression of nuclear Nrf2, which resulted in a significant increase of the ratio of nuclear Nrf2 to cytoplasmic Nrf2 (UUO7 vs. UUO7-FM, P = 0.012) (Fig. 3B).
3.1.5 Effects of fimasartan on Nrf2 downstream antioxidant genes and proteins
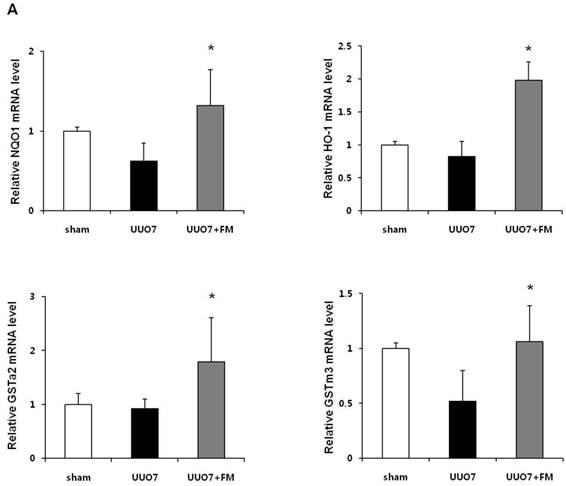
To examine the effect of fimasartan on the activation of Nrf2 downstream genes, the mRNA expression of NQO1, HO-1, GSTa2, and GSTm3 was measured. As shown in Figure 4A, UUO slightly decreased expression of the mRNA of these genes, but these decreases were not statistically significant as compared with those in the sham group. Treatment with fimasartan resulted in a significantly greater increase in the levels of mRNA of antioxidant genes, which showed that fimasartan induced transcription of these genes (UUO7 vs. UUO7-FM, P < 0.05).
Effect of fimasartan (FM) on the expression of Nrf2 downstream genes and proteins in unilateral ureteral obstruction (UUO). (A) The mRNA levels of NQO1, HO-1, GSTa2, and GSTm3. (B) Representative immunoblots of NQO1, HO-1, CuSOD, MnSOD, and catalase. (C) The immunofold of the expression of NQO1, HO-1, CuSOD, MnSOD, and catalase. UUO7 = UUO-control day 7; UUO7-FM = UUO-fimasartan day 7. *P < 0.05 vs. UUO7. †P < 0.05 versus sham and UUO7. Values are expressed as means ± SE.
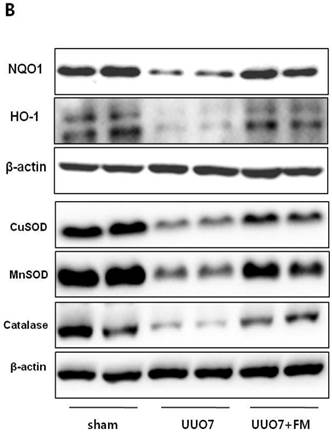
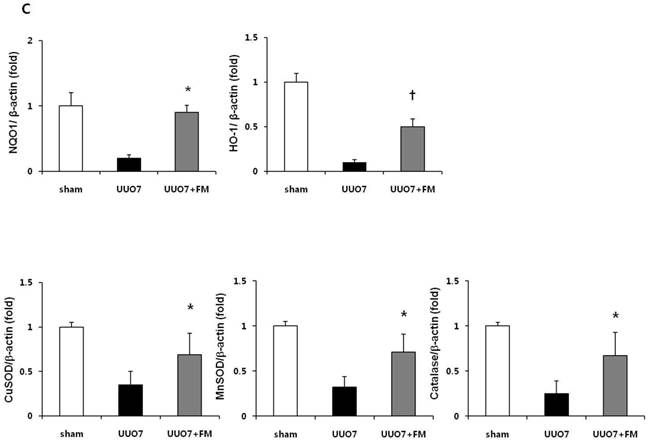
On immunoblot analysis, UUO prominently decreased the renal expression of Nrf2 downstream antioxidant enzymes including NQO1, HO-1, CuSOD, MnSOD, and catalase, as compared with that in the sham group (Fig. 4B). In agreement with the findings that fimasartan upregulates expression of the mRNA of NQO1 and HO-1, the protein expression of those genes, as well as of CuSOD, MnSOD, and catalase, was significantly increased in the fimasartan-treated UUO mice. (UUO7 vs. UUO7-FM: NQO1, P = 0.011; HO-1, P = 0.019; CuSOD, P = 0.021; MnSOD, P = 0.018; catalase, P = 0.038) (Fig. 4C).
3.1.6 Renal tubule cell apoptosis
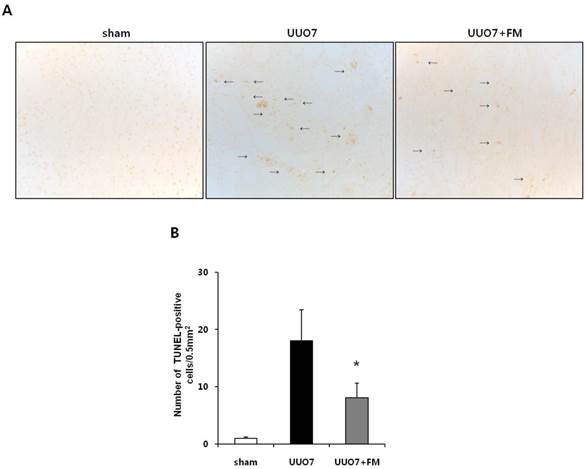
To determine the effects of fimasartan on oxidative stress-induced renal apoptosis, results of the TUNEL assay and the expression of pro- and anti-apoptotic markers were examined. As expected, the number of TUNEL-positive cells was increased in the UUO mice and was markedly decreased by treatment with fimasartan (UUO7 vs. UUO7-FM, 18.0±5.4 vs. 8.1±2.5 cells/0.5 mm2, P = 0.024) (Fig. 5A and 5B).
Effect of fimasartan (FM) on renal apoptosis in unilateral ureteral obstruction (UUO). (A) Representative photographs of TUNEL assay (×400). Black arrows indicate TUNEL-positive cells (brown color). (B) The number of TUNEL-positive cells in the obstructed kidney. (C) Representative immunoblots of Bax, Bcl-2, and Bim. (D) The ratio of the expression of Bax to that of Bcl-2 and Bim. UUO7 = UUO-control day 7; UUO7-FM = UUO-fimasartan day 7. *P < 0.05 vs. sham and UUO7. †P < 0.05 vs. UUO7. Values are expressed as means ± SE.
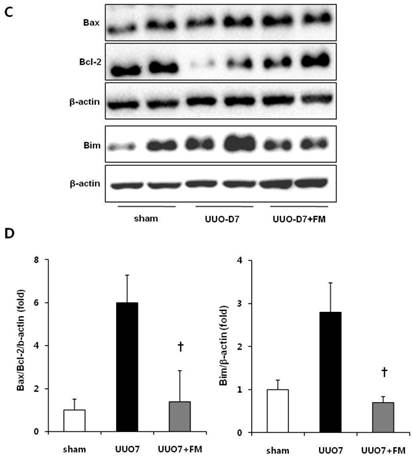
UUO mice showed slightly increased expression of the pro-apoptotic gene Bax, along with markedly decreased expression of the anti-apoptotic gene Bcl-2. Fimasartan treatment induced the increased expression of Bcl-2, which resulted in a significant decrease in the ratio of Bax to Bcl-2 (UUO7 vs. UUO7-FM, P = 0.041). The expression of Bim, another pro-apoptotic gene, was also elevated in the UUO mice. The fimasartan treatment attenuated these UUO-induced changes, indicating that such treatment is indeed effective in preventing UUO-induced apoptosis (UUO7 vs. UUO7-FM, P = 0.028) (Fig. 5C and 5D).
3.2. Effects of fimasartan in HK-2 cells
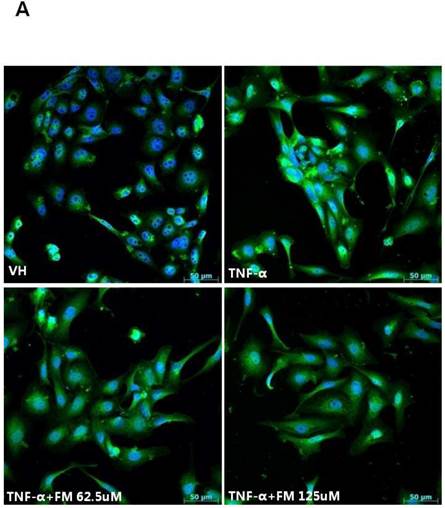
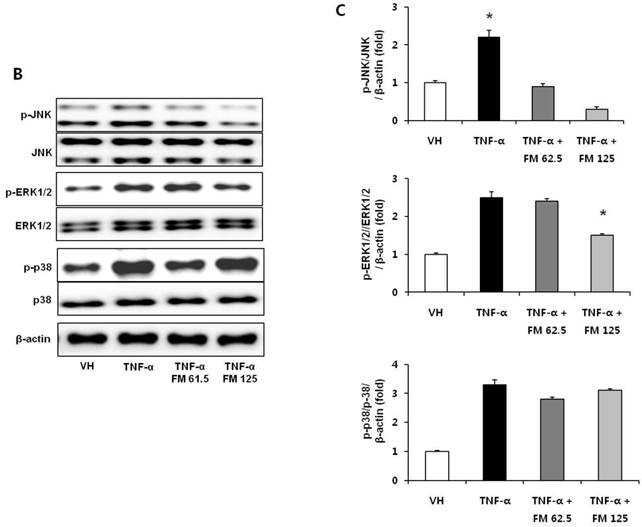
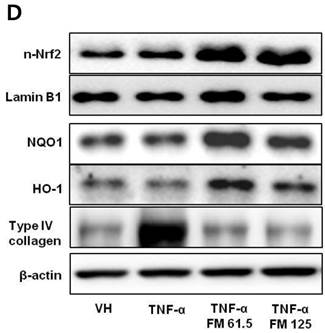
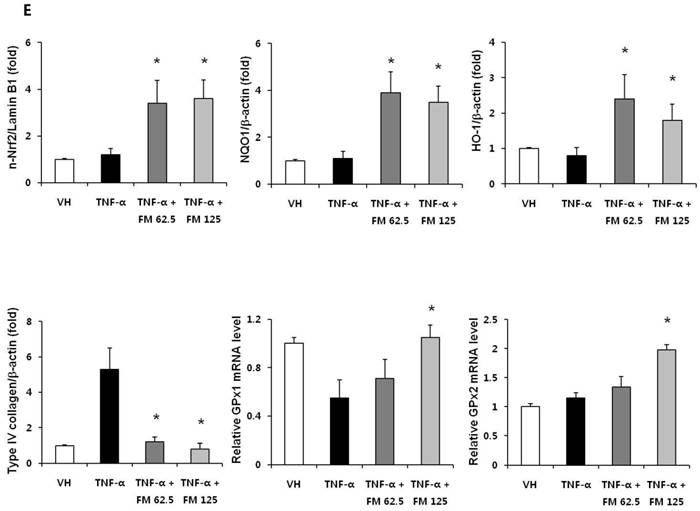
In HK-2 cells, exposure to TNF-α (5 ng/ml) prominently increased the number of ATII-positive cells, but this number was decreased by pretreatment with fimasartan (at concentrations of 62.5 and 125 µM) (Fig. 6A). The protein levels of phospho-JNK, phospho-ERK1/2, and phospho-p38 MAPK were significantly elevated in the HK-2 cells exposed to TNF-α. Pretreatment of cells with fimasartan inhibited phosphorylation of JNK and ERK1/2 but not p38 MAPK. (Fig. 6B and 6C). Compared with vehicle, TNF-α stimulation did not change the expression of nuclear Nrf2; however, it was markedly increased in TNF-α-stimulated HK-2 cells following pretreatment with fimasartan (Fig. 6D and 6E). Consistent with the findings in renal tissues of mice, the expression of Nrf2 downstream antioxidant enzymes including NQO1 and HO-1 was significantly increased in the fimasartan pretreatment group (Fig. 6D and 6E). Consequent increased expression of Nrf2 downstream targets was observed in mRNA levels of GPx1 and GPx2 genes (TNF-α vs. TNF-α + 125 µM, P < 0.05) (Fig. 6E). TNF-α stimulation prominently increased the expression of type IV collagen, the degree of which was significantly attenuated by fimasartan pretreatment (Fig. 6D and 6E).
Immunofluorescence analysis to show ATII expression, immunoblot p-JNK, nuclear Nrf2 and antioxidant enzymes, and the mRNA levels of nuclear Nrf2 downstream genes in HK-2 cells. (A) Representative photographs of ATII expression. Scale bar = 50 µM. (B) Representative immunoblots of p-JNK, p-ERK1/2, and p-38 MAPK. (C) The immunofold of the expression of p-JNK, p-ERK1/2, and p-38 MAPK. (D) Representative immunoblots of nuclear Nrf2, NQO1, HO-1, and type IV collagen. (E) The immunofold of the expression of nuclear Nrf2, NQO1, HO-1, and type IV collagen, and the mRNA levels of GPx1 and GPx2. VH = vehicle; FM = fimasartan. *P < 0.05 vs. TNF-α.
Discussion
In this study, renal injury was induced by unilateral ureteral obstruction in mice, as evidenced by renal tissue inflammation and fibrosis, as well as RAS activation, decreased expression of antioxidant enzymes, and increased apoptosis. Treatment with fimasartan had a protective effect against UUO-induced renal injury by reducing RAS activation and oxidative stress, and renal inflammation and fibrosis were consequently reduced. The level of SBP of the mice in all three groups was similar before sham or UUO surgery and was maintained throughout the experiment. Fimasartan did not lower the SBP of mice at a dosage of 3 mg/kg/day, suggesting that the salutary effect of fimasartan on renal injury may be due to its anti-inflammatory or antifibrotic properties, not its hemodynamic function.
In recent years, evidence has accumulated showing that ATII raises arterial pressure, causes renal tissue inflammation, and extracellular matrix protein accumulation, which results in the progression of renal fibrosis [16]. Binding of ATII to AT1R induces the activation of NADPH oxidase, which co-localizes with AT1R in the cell membrane [17]. NADPH oxidases (Noxs) are distributed throughout the kidney, including the renal vessels, glomeruli, tubules, and interstitium. Renal vessels and proximal tubules express Nox1, Nox2, and Nox4, and such expression can be upregulated in diabetes as well as in hypertension. These Noxs are known to produce superoxide as their primary enzymatic product [18]. Previous studies have shown that Nox activation mediated by AT1R results in the generation of ROS [19]. Tian et al. found that Nox contributes to oxidative stress and inflammation in the renal tissue of rats, as well as to renal injury and dysfunction [20].
In our study, the renal expression of AT1R was increased in the UUO mice, and treatment with fimasartan led to the downregulation of AT1R. We also measured renal expression of Nox1, Nox2, and Nox4 by immunoblot analysis. The results revealed that UUO significantly increased Nox expression levels, and such expression was suppressed by treatment with fimasartan. In addition, we showed that UUO induced apoptosis, which would be expected to occur through the generation of ROS, but this effect was significantly ameliorated with fimasartan treatment, as evidenced by changes in the number of TUNEL-positive cells and the expression levels of pro-apoptotic or anti-apoptotic genes.
Nrf2 is known to be a critical transcription factor that binds to the ARE in the promoter region of several genes encoding for antioxidant enzymes in cells and tissues [21-23]. It induces intracellular antioxidants and phase II detoxifying enzymes such as NQO1, HO-1, glutathione S-transferase (GST), glutamate-cysteine ligase, and glutathione peroxidase (GPx) [24]. The induction of these proteins, which is referred to as an Nrf2-mediated defense response, is critical for neutralizing ROS and for cell survival in counteracting the adverse effects of various exogenous insults. ATII is also known to generate ROS in renal mesangial cells and to inhibit the basal protein expression of Nrf2 and its downstream antioxidant enzymes [25]. A previous experimental study of rats with spontaneous focal glomerulosclerosis demonstrated that ARB treatment restored Nrf2 activation and expression of the detoxifying antioxidant enzymes, which led to the suppression of ROS, inflammation, and apoptosis in renal tissue [8].
Recently, it has been reported that telmisartan suppressed NADPH oxidase and upregulated Nrf2, which decreased renal oxidative stress [26]. Although in our study the expression of nuclear Nrf2 did not decrease after UUO, fimasartan significantly increased the level of nuclear Nrf2. UUO markedly decreased the expression of cytoplasmic Nrf2, and treatment with fimasartan did not change these cytoplasmic levels. These results indicate that fimasartan may facilitate dissociation of the Keap1-Nrf2 complex and translocation of Nrf2 to the nucleus. Consistent with previous reports, our study demonstrated that activation of Nrf2, as evidenced by increased nuclear translocation of Nrf2, led to the upregulation of Nrf2 downstream target genes that encode phase II detoxifying enzymes. In addition to gene expression changes in mRNA levels, immunoblot analysis revealed a significant increase in the expression of antioxidant proteins, including CuSOD, MnSOD, and catalase, in the fimasartan-treated mice.
Excessive accumulation of extracellular matrix proteins and fibrosis are the major pathologic features of UUO-induced nephropathy and contribute to the progression of chronic renal failure [27]. It has been reported that TNF-α-induced alterations in matrix metalloproteinases and tissue inhibitors of metalloproteinases promote renal tubulointerstitial fibrosis, and the MAPK signaling pathway may be involved in this process [28, 29]. Furthermore, the MAPKs play a critical role in ATII-induced renal interstitial fibrosis, and blockade of RAS may contribute to the prevention of renal interstitial fibrosis [30]. The Nrf2 signaling pathway can be activated by upstream kinases including MAPKs, protein kinase C, and phosphatidylinositol-3-kinase/Akt [31]. Our findings suggest that in TNF-α-stimulated HK-2 cells, phospho-JNK and phospho-ERK1/2, but not phospho-p3 MAPK, may be involved in the process of Nrf2 activation following pretreatment with fimasartan.
In conclusion, progressive renal inflammation and fibrosis in mice in the UUO group was associated with the upregulation of oxidative stress and remarkable impairment of Nrf2 activation. Treatment with fimasartan prevented the development of nephrotoxicity by reducing oxidative stress, inflammation, and fibrosis in renal tissues. Fimasartan induces amelioration of oxidative stress, subsequently lowering ROS production and increasing the production of endogenous antioxidant enzymes. The possible renoprotective mechanisms of fimasartan may include the upregulation of the Nrf2 signaling pathways induced by suppression of RAS activation and inhibition of the phospho-JNK and phospho-ERK1/2 pathway in this experiment. In spite of the positive results presented here, we could not fully demonstrate a clear mechanism to explain the effects of fimasartan. Therefore, further studies are needed to determine the pharmacologic actions of fimasartan associated with the activation of Nrf2 in this UUO model.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Satirapoj B, Adler SG. Comprehensive approach to diabetic nephropathy. Kidney Res Clin Pract. 2014;33:121-31
2. Himmelfarb J. Linking oxidative stress and inflammation in kidney disease: which is the chicken and which is the egg? Semin Dial. 2004;17:449-54
3. Dendooven A, Ishola DA Jr, Nguyen TQ. et al. Oxidative stress in obstructive nephropathy. Int J Exp Pathol. 2011;92:202-10
4. Percy C, Pat B, Poronnik P, Gobe G. Role of oxidative stress in age-associated chronic kidney pathologies. Adv Chronic Kidney Dis. 2005;12:78-83
5. Truong LD, Gaber L, Eknoyan G. Obstructive uropathy. Contrib Nephrol. 2011;169:311-26
6. Reisman SA, Aleksunes LM, Klaassen CD. Oleanolic acid activates Nrf2 and protects from acetaminophen hepatotoxicity via Nrf2-dependent and Nrf2-independent processes. Biochem Pharmacol. 2009Apr1;77(7):1273-82
7. Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol. 2010;298:F662-71
8. Kim HJ, Sato T, Rodríguez-Iturbe B. et al. Role of intrarenal angiotensin system activation, oxidative stress, inflammation, and impaired nuclear factor-erythroid-2-related factor 2 activity in the progression of focal glomerulosclerosis. J Pharmacol Exp Ther. 2011;337:583-90
9. Li W, Khor TO, Xu C. et al. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol. 2008;76:1485-9
10. Sachse A, Wolf G. Angiotensin II-induced reactive oxygen species and the kidney. J Am Soc Nephrol. 2007;18:2439-46
11. Remuzzi G1, Perico N, Macia M. et al. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney Int Suppl. 2005;99:S57-65
12. Chi YH, Lee H, Paik SH. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of fimasartan following single and repeated oral administration in the fasted and fed states in healthy subjects. Am J Cardiovasc Drugs. 2011;11:335-46
13. Lee JY, Lee CW, Kim WJ. et al. Antiatherosclerotic effects of the novel angiotensin receptor antagonist Fimasartan on plaque progression and stability in a rabbit model: a double-blind placebo-controlled trial. J Cardiovasc Pharmacol. 2013;62:229-36
14. Zhang G, Kim H, Cai X. et al. Urokinase receptor deficiency accelerates renal fibrosis in obstructive nephropathy. J Am Soc Nephrol. 2003;14:1254-71
15. Chung S, Yoon HE, Kim SJ. et al. Oleanolic acid attenuates renal fibrosis in mice with unilateral ureteral obstruction via facilitating nuclear translocation of Nrf2. Nutr Metab. 2014;11:2
16. Hoffmann S, Podlich D, Hähnel B. et al. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol. 2004;15:1475-87
17. Garrido AM1, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 2009;302:148-58
18. Holterman CE, Read NC, Kennedy CR. Nox and renal disease. Clin Sci. 2015;128:465-81
19. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82-97
20. Tian N, Moore RS, Phillips WE. et al. NADPH oxidase contributes to renal damage and dysfunction in Dahl salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1858-65
21. Dhakshinamoorthy S, Jaiswal AK. Small maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:quinone oxidoreductase1 gene. J Biol Chem. 2000;275:40134-41
22. Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549-57
23. Kwak MK1, Wakabayashi N, Kensler TW. Chemoprevention through the Keap1-Nrf2 signaling pathway by phase 2 enzyme inducers. Mutat Res. 2004;555:133-48
24. Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769-89
25. Kang KW, Lee SJ, Kim SG. Molecular mechanism of nrf2 activation by oxidative stress. Antioxid Redox Signal. 2005;7:1664-73
26. Fujita H, Fujishima H, Morii T. et al. Modulation of renal superoxide dismutase by telmisartan therapy in C57BL/6-Ins2(Akita) diabetic mice. Hypertens Res. 2012;35:213-20
27. Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75:1145-52
28. Nee LE, McMorrow T, Campbell E. et al. TNF-alpha and IL-1beta-mediated regulation of MMP-9 and TIMP-1 in renal proximal tubular cells. Kidney Int. 2004;66:1376-86
29. Cha JJ, Hyun YY, Jee YH. et al. Plasma leptin concentrations are greater in type II diabetic patients and stimulate monocyte chemotactic peptide-1 synthesis via the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway. Kidney Res Clin Pract. 2012;31:177-85
30. Sekine S1, Nitta K, Uchida K. et al. Possible involvement of mitogen-activated protein kinase in the angiotensin II-induced fibronectin synthesis in renal interstitial fibroblasts. Arch Biochem Biophys. 2003;415:63-8
31. Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008;74:1526-39
Author contact
![]() Corresponding author: Seok Joon Shin. Address: 403-720 #56 Dongsuro, Bupyeong-gu, Incheon, Korea. Phone: 82-32-280-5091; Email: imkidneyac.kr
Corresponding author: Seok Joon Shin. Address: 403-720 #56 Dongsuro, Bupyeong-gu, Incheon, Korea. Phone: 82-32-280-5091; Email: imkidneyac.kr