Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Role of pirfenidone in the...
Lung
Effect of pirfenidone in IPF
Adverse effects of pirfenidone
Other drugs for IPF treatment
Kidney
Heart
Eye
Liver
Role of PFD in the regulation of...
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(11):840-847. doi:10.7150/ijms.11579 This issue Cite
Review
Role and New Insights of Pirfenidone in Fibrotic Diseases
David Alejandro Lopez-de la Mora1, Cibeles Sanchez-Roque1, Margarita Montoya-Buelna1, Sergio Sanchez-Enriquez1, Silvia Lucano-Landeros1, Jose Macias-Barragan1,2, Juan Armendariz-Borunda1, ![]()
1. Institute for Molecular Biology and Gene Therapy, Department of Molecular Biology and Genomics, University of Guadalajara, Sierra Mojada St. 950, Guadalajara (44280), Mexico.
2. Departamento de Ciencias de la Salud, CUValles, University of Guadalajara, Guadalajara - Ameca km. 45.5, Ameca (46600), Mexico.
Received 2015-1-12; Accepted 2015-7-16; Published 2015-10-14
Abstract
Pirfenidone (PFD) is a non-peptide synthetic molecule issued as a broad-spectrum anti-fibrotic drug with the ability to decrease TGF-β1, TNF-α, PDGF and COL1A1 expression, which is highly related to prevent or remove excessive deposition of scar tissue in several organs. Basic and clinical evidence suggests that PFD may safely slow or inhibit the progressive fibrosis swelling after tissue injuries. Furthermore, a number of evidence suggests that this molecule will have positive effects in the treatment of other inflammatory diseases. This review contains current research in which PFD has been used as the treatment of several diseases, and focus mainly in the outcomes related to improve inflammation and fibrogenesis. Therefore, the main goal of this review is to focus on the novel findings of PFD efficacy rather than deepen in the chemical aspects of the molecule.
Keywords: Pirfenidone, fibrosis, inflammation, idiopathic pulmonary fibrosis
Introduction
This review intends to draw the attention of the reader on the updated and current knowledge of the use of Pirfenidone (PFD) in the treatment of several fibrotic diseases. It is structured in such a way that the reader will be able to grasp easily the actual concept on PFD action on different organs. Each section is divided in the basic preclinical studies, followed by the most recent findings in the clinical scenario.
PFD is being investigated for therapeutic profits to patients suffering from fibrosis conditions such as Hermansky-Pudlak Syndrome (HPS), associated pulmonary fibrosis and idiopathic pulmonary fibrosis (IPF). PFD has also been studied to analyse pharmacologic effects in preventing, or even stopping, scarring process found in fibrosis in injured tissues including those of lungs, skin, joints, kidneys, prostate glands and liver [1].
Research suggests that PFD may safely slow or inhibit the progressive enlargement of fibrotic lesions, and prevent formation of new lesions following tissue injuries. PFD is provided for oral administration in capsules or tablets. Different formulations have been tested and implemented in clinical trials, additional research and experiments. There are a number of reviews available regarding the action and pharmacokinetics of PFD [2-5]. The main goal of this review is to focus on the novel findings of PFD efficacy rather than deepen in the chemical aspects of this molecule.
Role of pirfenidone in the regulation of fibrosis
Fibrosis is the state resulting from excessive accumulation of extracellular matrix components such as collagen and fibronectin secreted by myofibroblasts in response to cellular damage. Parenchyma replacement by extracellular matrix excess during chronic fibrosis damage eventually leads to organ failure [2,5]. In addition, fibrosis is a condition arising from chronic state of various diseases such as scleroderma, rheumatoid arthritis, Crohn's disease, ulcerative colitis, systemic lupus erythematosus and idiopathic pulmonary fibrosis (IPF). These events fall into three main mechanisms: cell damage, inflammation and eventually fibrosis. These pathologies cause organ dysfunction, through permanent scar tissue accumulation, and many even lead to death [6,7]. PFD has been tested as an anti-fibrotic drug in lung, liver, kidney and myocardium, although other studies have proposed it as a therapeutic adjunct in the prevention of allografts [8]. It has also been shown that PFD reduces keloid formation in an animal model and our own clinical data has demonstrated that a scar reduction with 8% PFD gel (KitoscellTM; Cell Therapy and Technology, Mexico City, Mexico) topically administered during a 6-month period, led to resolution of hypertrophic scars acquired after burns in pediatric patients [2].
Lung
The use of PFD in lung tissue has been extensively studied, which has led to describe several therapeutic targets, especially those related to anti-inflammatory and anti-fibrogenic actions directing attention to research development in conditions that compromise pulmonary tissue architecture, mainly in IPF. This chronic and progressive disease is characterized by a progressive decline in lung function. The prevalence of IPF is estimated at 20/100,000 for males and 13/100,000 for females with a greater incidence in individuals aged over 50 years; approximately two-thirds are over the age of 60 years old at the time of disease beginning [9,10]. The etiology of IPF remains unclear and the only risk factors associated with the disease onset are smoking, exposure to toxic gases, genetics and environmental factors [11-17].
IPF pathophysiology has not been fully defined but is attributed to organ damage, specifically, epithelial damage with consequent activation of alveolar cells that secrete a cascade of pro-inflammatory molecules activating fibroblast proliferation and myofibroblast differentiation with a reduced apoptosis rate. Therefore, the main therapeutic target for IPF is the decrease of extracellular matrix accumulation [11-17]. IPF is characterized by irregular scattered fibrotic areas, fibroblast proliferation and epithelial to mesenchymal transition, where cells in turn are the major source of profibrogenic mediators such as platelet derived growth factor (PDGF), fibroblast growth factor (FGF) and TGF-β1, which has been shown to increase mRNA expression of collagen type I and heat shock protein 47 (HSP47), a collagen-specific chaperone, located in the endoplasmic reticulum which is engaged in processing, assembly and secretion of procollagen during fibrotic processes.
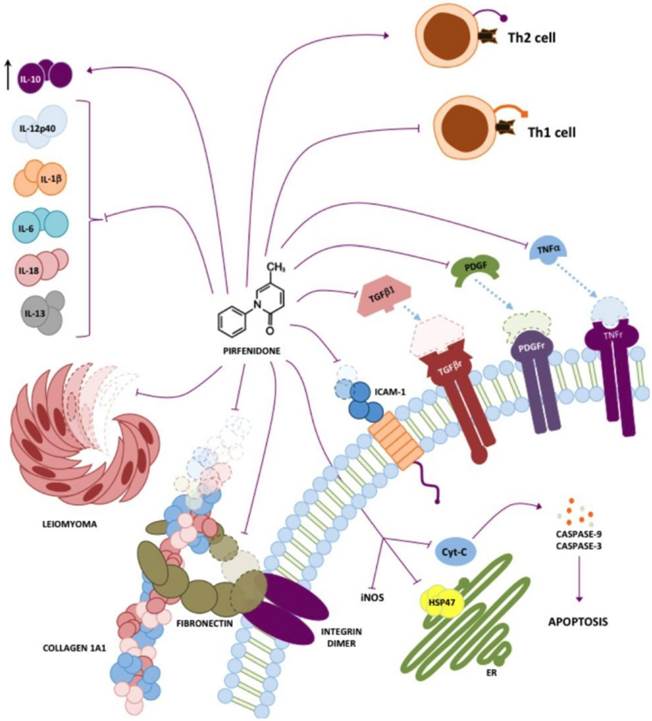
It has also been demonstrated that PFD inhibits TGF-β1-induced over-expression of collagen type I, and heat shock protein 47 in A549 cells. These mechanisms are summarized in figure 1 [13].
Pirfenidone targets. Different targets in vivo and in vitro for PFD have been described, the most prominent being inhibition of TGF-β1 and TNF-α. However, it has also been shown that PFD has either direct or indirect action on other molecules such as collagen I, PDGF, IL-6, IL-1β, IL-13, IL-12p40, fibronectin, HSP47 and ICAM-1.
Effect of pirfenidone in IPF
PFD has been tested in experimental and clinical models of IPF with favorable results. In phase II and III clinical trials, PFD has shown to improve the decrease of functional vital capacity (FVC) of patients at 32 and 52 weeks respectively up to 44% compared to placebo, improving survival (p=0.0280) rate. This phase III trial, has shown that basal FVC ≥ 70 % and oxygen saturation < 90 % (measured by pulse oximetry (SpO2)) during the 6MET, low doses of PFD (1800 and 1200 mg/day) provide more benefits to patients in terms of FVC and progression- free survival (PFS) changes, but also in subjective symptoms such as cough and dyspnea compared to placebo patients [10]. Studies as CAPACITY 004 and 006 have also shown reduction in FVC decline at 72th week of treatment, improving parameters in the test results of 6-minute walk test (6MWT) at the standard dose of 2403 mg/day [18].
In other multicenter phase III trial, the efficacy of PFD was analyzed in patients with IPF and results shown in this trial suggested that, when 5 % change in VC was used as an index instead of the 10 % change, the efficacy of PFD could be evaluated with higher sensitivity and robustness over the 12-month study. It was also suggested that 5 % change in FVC at month 3 is clinically useful and a significant promising prognostic factor of IPF [19].
Systematic reviews has been conducted in order to investigate effectiveness of available IPF treatments, Loveman E (2015) analyzed fourteen studies of clinical effectiveness, one evaluated azathioprine, three N-acetylcysteine (NAC) (alone or in combination), four pirfenidone, one BIBF 1120, one sildenafil, one thalidomide, two pulmonary rehabilitation, and one a disease management program. Authors observed that these studies were generally good, with low risk of bias, and that some treatments appear to be clinically effective. The model base-case results showed increased survival for five pharmacological treatments, but general recommendations cannot be made due to cost-effectiveness and to limitations in the evidence base [20].
Also, a phase II study (WJOG 6711L) has been developed to validate effectiveness and the safety of perioperative administration of PFD to lung cancer patients with IPF, trying to analyse whether PFD reduces lung injury after lung resection [21].
Adverse effects of pirfenidone
In the vast majority of the clinical protocols carried out to date, the most common adverse effects of PFD have been shown to be photosensitivity and rash as dermatological issues; gastrointestinal: nausea, diarrhea, and dyspepsia; neurological: fatigue, insomnia and dizziness [22].
It is important to notice the efforts of Onoue et al. in the sense to device an alternative delivery of PFD as an inhalable powder. By using this pharmaceutical preparation they reduced phototoxicity in an experimental model with favorable results. This kind of findings calls for the design of new forms of PFD delivery to reduce sensitivity and dosing required achieving therapeutic levels increasing bioavailability in nasal mucosa [23]. Eventually, this type of strategy will have to be assayed in human beings under the scrutiny of controlled clinical trials. However, more studies should be carried out since treatment effectiveness is related to the time and concentration of the drug at the site of damage. In the case of IPF, PFD would have to be not only in the nasal mucosa but reach the alveoli for better diffusion in the lung tissue.
Another trial named CAPACITY, rendered data that only 1% of patients discontinued treatment due to nausea and another 1% due to rash [10,11]. The extension phase of the study CAPACITY named RECAP, evaluated the safety of PFD after phase III studies to confirm the tolerability of PFD in patients who were treated for a mean of 2.9 years [12] though safety and tolerability of PFD treatment has been questioned by other authors [21,24]. On the other hand, a prospective, double-blind, randomized, placebo-controlled trial that included 35 patients with type 1 Hermansky-Pudlak syndrome (HPS-1), investigated the safety and efficacy of PFD in mild to moderate HPS-1 (NHGRI protocol 97-HG-0085). During the trial there were 10 severe adverse events (SAE) and several non-serious adverse events including 3 deaths, considered to be part of the natural course of the lung disease, chest pain (2 subjects), elevated CPK (2 subjects), deep vein thrombosis, hematochezia and otitis with vertigo on one subject each one. Furthermore, the most commonly reported non-serious adverse events include dyspepsia and heartburn in approximately 50% of the subjects in placebo and PFD groups, and almost all patients required anti-acid therapy. Other adverse events included photosensitivity rash. There was no evidence of bone marrow (hemoglobin, total leucocyte count), renal (serum creatinine), hepatic toxicity (alanine aminotransferase) or cardiac toxicity [25,26].
Among the broad spectrum of PFD actions, it has been suggested the use of this molecule to treat pulmonary fibrosis in patients previously poisoned by mustard gas, since the intermediate products originated by this molecule tend to permanently alkylate guanine from DNA, preventing cellular division and inducing cell death. Tissues exposed to this agent could present side effects even years after gas exposure, developing for example abnormal skin pigmentation, eye problems, cancer and pulmonary fibrosis. Therefore, the use of PDF could be helpful to treat exposed soldiers or workers to this hazardous material in the attempt to reduce signs and symptoms of pulmonary fibrosis [27].
Other drugs for IPF treatment
Some therapies have been developed to treat IPF, such as corticosteroids, azathioprine, azathioprine and prednisolone, cyclophosphamide, everolimus, anticoagulants, endothelin inhibitors I (ET), Sildenafil, Interferon, Etanercept, Imatinib, CC-930, N-acetylcysteine (NAC), monoclonal antibodies, angiotensin receptor blockers as losartan A1, somatostatin analogues such as Octreotide, etc. [15,16,24,28]. However some of these strategies did not produce significant benefits, or even were more harmful for disease treatment. Besides the side effects of different therapies, correct stratification of patients according to degree of pulmonary function is a limitation that reduce the application of specific dose of many drugs, for example the triple therapy with NAC, azathioprine and prednisone, which unfortunately have shown an increased death rate and hospitalization compared with placebo [12,15]. However other therapies such tyrosine kinase inhibitors as BIBF 1120 (Nintedanib) have been approved for application in patients with IPF in January 2015 based on results from the replicate Phase III INPULSIS trials. This therapy slowed disease progression by reducing the annual rate of decline in lung function by 50% in a broad range of IPF patients [29].
Regardless of the armamentarium available as potential candidates to be used to treat this life-threatening disease, the relatively positive results in various tests of lung function enabled PFD as one of the main approved drug to treat mild to moderate IPF in Japan, China and India in 2008 [21] and in February 2011 it was also approved by the European Commission for the treatment of the same lung condition.
The ASCEND clinical trial (PIPF-016) was a randomized, double-blind, placebo controlled, phase 3 study of the efficacy and safety of PFD in patients with IPF aiming to confirm the PFD-treatment effect on change in predicted forced vital capacity (%FVC), and to confirm the safety of PFD treatment. This clinical trial enrolled 555 patients from 127 sites in 9 countries, mostly in the U.S., 278 were assigned to receive PFD, and 277 were assigned to receive placebo. Subjects had clinical-radiographic or biopsy-confirmed IPF, and mild to moderate restrictive lung disease (FVC, forced vital capacity 50-90% predicted) with diffusion impairment on pulmonary function testing, and were randomized 1:1 to PFD or placebo one-year treatment. After 52 weeks, patients receiving PFD had a significant decline in FVC (≥10% predicted) compared to placebo patients (16.5% vs. 31.8%), which emerged early and increased during the course of the trial. Also, the 6-minute walk distance test increased 27 m (Relative difference 44.2%; P=0.04) in subjects taking PFD. The highly significant finding was supported by the encouraging effect on rates of death from any cause (4.0% vs. 7.2%) and from IPF (1.1% vs. 2.5%), since progression-free survival (PFS) was reduced with PFD (HR 0.57; 95% CI 0.43-0.77; P<0.001). As mentioned before in many clinical trials, it is difficult to reconcile results obtained from tested groups with all established stages in a disease, since these observations were obtained from enrolled patients with mild-to-moderate physiological impairment, results from population of patients with advanced disease are therefore uncertain. Based on this evidence authors conclude that PFD as compared with placebo reduced disease progression in patients with IPF, treatment was generally safe with an acceptable side-effect profile, and was associated with fewer deaths by slowing the rate of IPF worsening. Therefore, evidence shown in this research is positive for patients with IPF who have a very poor prognosis [18]. Based in this evidence, PFD was approved by FDA for use in IPF [30].
Kidney
Renal fibrosis is a common pathway resulting from chronic renal damage leading to end-stage renal disease (ESRD) and organ dysfunction. Renal fibrosis is a dynamic, interactive process initiated by cellular damage, characterized by loss of cellular differentiation phenotype and accumulation of extracellular matrix proteins. Any type of cell damage leading to secretion of pro-inflammatory cytokines and its consequent chronic inflammatory process culminates in a fibrotic process [30]. The treatment of renal fibrosis is limited and largely ineffective because many patients progress to diabetic nephropathy, which is one of the most common complications of diabetes mellitus, and progress to ESRD. PFD has been show to significantly reduce renal fibrosis in several pilot studies. However more research is needed to understand in a comprehensive manner the mechanism, indications and contraindications in appropriate candidate patients [31,32]. 77 patients with diabetes reduced glomerular filtration rate (eGFR) and proteinuria were included randomly into three groups (placebo, 1,200 mg of PFD and 2,400 mg of PFD daily) of study. After 54 weeks of treatment, 1,200 mg of PFD treated group, improved eGFR with an increase of 3.3 ml/min/1.73 m2. Group treated with 2,400 mg of PFD group achieved mild eGFR change, which was not significant compared to placebo. However, urine TGF-β1 protein levels measured by ELISA were not significantly affected by PFD. Therefore, this results show that PFD is a drug that can prevent renal failure and even improve renal function in patients with diabetic nephropathy [32,33,34].
Heart
In heart disease, there are several ailments that can lead to fibrosis development and cardiac complications, such conditions include hypertension, myocardial infarction and heart failure, which could constitute stimulus for causing cardiac fibroblasts proliferation, transformation into myofibroblasts, extracellular matrix synthesis and migration with consequent fibrosis driving to complications as atrial fibrillation (AF). It has been shown in cultured cardiac fibroblasts that PFD inhibits α-SMA expression this molecule is involved in morphologic transformation of fibroblasts into myofibroblasts, proliferation, migration and secretory capacity of these cells. Moreover, PFD inhibits the expression of extracellular matrix metalloproteinase MMP-9 and increases expression of TIMP-1 and IL-10 while reducing the synthesis and secretion of TGF-β1 [35,36], these features have been associated with the establishment of AF in subjects with essential hypertension, since these mediators are highly associated to the onset of atrial fibrosis which in term, could be lowered with PFD administration [37].
Furthermore, PFD could be used as an antifibrotic treatment for ventricular hypertrophy secondary to hypertension, which has been shown in an experimental model of left ventricular hypertrophy induced by angiotensin II, it was observed that PFD treatment reduce heart weight and decreased left ventricular hypertrophy, inhibits perivascular and interstitial tissue fibrosis, and reduce atrial natriuretic peptide, brain natriuretic peptide and TGF-β1 mRNA gene expression in mice cardiac tissue [38].
Different studies have been conducted to analyse PFD effect to treat Hypertrophic Cardiomyopathy (NCT00011076) and Heart Function and Exercise Capacity in Patients With Hypertrophic Cardiomyopathy (NCT00354328), but no information regarding outcomes of these trials have been published yet. Whether it was the case that such protocols were unsuccessful, even so results should be published, so it would greatly reduce speculation about drug effects in this ailments [39,40].
Eye
As described in other organs in which PFD had an inhibitory effect on proliferation, migration and collagen synthesis by fibroblasts, human orbital fibroblasts (Tenon capsule fibroblast) are not exempted from these effects. Zhong et al. tested in a rat model, topical PFD as an antifibrotic postoperative treatment for glaucoma filtration surgery (such as trabeculotomy), since postoperative scar represents the major barrier in the long-term control intraocular pressure since it can block the exit (outflow) of the aqueous humor, leading to optic nerve damage. In this study, favorable results were obtained at a dose of 0.5% of PFD, groups with PFD and reference (mitomycin C, MMC) treatment developed mild fibrosis and collagen deposition in subconjunctival space. However, it is known that MMC has some degree of toxicity, therefore PFD based treatment is a promise in preventing postoperative scar formation, since PFD showed no toxicity in cornea, retina ciliary body, or may be a safer drug than MMC. Subsequent studies have shown that therapeutic doses can be achieved up to 2 hours, since PFD can be internalized into conjunctiva, and vitreous and aqueous humor, sclera and cornea after topical administration, opening the door for future research to evaluate whether PFD is useful in diabetic retinopathy and proliferative vitreoretinopathy [41,42].
A common eye disease is cataract lens opacity which conventional treatment is surgery, however posterior camera opacity (PCO) complication due to fibrosis is common after excision. PFD has been tested in vitro as a possible treatment for PCO prevention. Previous studies have shown that PFD prevented fibrogenesis by inhibiting nuclear translocation of Smad 2/3, which in term regulates TGF-β1 protein phosphorylation resulting in down-regulation of mesenchymal markers in human retinal pigment epithelial cell line (ARPE-19) [43], beside these observations PFD also inhibits cell proliferation, migration and epithelial-mesenchymal transition (EMT) in human lens epithelial cell line SRA01/04 (HLECs) [44]. Another study has compared PFD antifibrotic effects with dexamethasone in cultured fibroblasts isolated from thyroid-associated ophthalmopathy (TAO) patients subjected to orbital decompression surgery, PFD proved to be more effective than dexamethasone in reducing TIMP-1 protein measured by ELISA and hydroxyproline in orbital fibroblasts of TAO patients. Thus, PFD might represent a promise as future antifibrotic agent in thyroid ophthalmopathy [45].
Liver
Evidence of PFD treatment in humans with liver injury is limited. Nonetheless, our own studies [46,47] have shown that PFD might be of use in the treatment of established advanced liver fibrosis caused by chronic infection with hepatitis C virus. We have carried out two different protocols, which have rendered promising data on the use of this antifibrotic drug in a functional compromised liver.
Chronic hepatitis C (CHC) is one of the most common etiologies for liver fibrosis and will eventually progress to cirrhosis or, in a less extent, to hepatocellular carcinoma. Nowadays, it is estimated that up to 3% of the world population is affected by CHC, thus, a great deal of drugs designed to clear the liver from the infectious viral component in this disease, have been developed. Nevertheless, fibrotic sequels eventually leading to dysfunctional liver activity in these patients are far from being resolved. It is well known that PFD has proved anti-fibrotic and anti-inflammatory properties in a wide number of animal models of fibrosis. PFD effects are mediated in part through inhibition of NF-κB protein activation [48], reduction of TNF-α and IFN-α levels and decrease in iNOS/NO induction in the liver of rats [49]. Also, PFD down-regulates TGF-β1, TIMP-1, MMP-2 mRNA and collagen deposition in a rat experimental model of cirrhosis induced by CCl4 [50,51].
Previously, our group demonstrated that 12-month treatment with 1200 mg/day of oral PFD in patients with established liver fibrosis, decreased liver necroinflammation, steatosis and at less extent, fibrosis. Liver collagen I, TGF-β1 and TIMP-1 mRNAs were also down-regulated [46]. Recently, we have reported data of an additional study aimed to assess a 24-month treatment with PFD and its influence in necroinflammation, fibrosis and steatosis, serum levels of TGF-β1, IL-6, TNF-α and liver CB1 and CB2 gene expression, in CHC patients. Twenty-eight patients out of 34 with CHC virus infection were enrolled in the study and received PFD (1200 mg/day) for 24 months. Liver biopsies at the end of treatment demonstrated that necroinflammation grading was reduced in an average of 3.2 points in 82% of patients (p < 0.05) and Ishak's fibrosis stage decreased 2-points average in 67% of patients (p < 0.05). Serum samples did show that IL-6, TGF-β1 and TNF-α levels decreased significantly in 93%, 67% and 47% of patients respectively and increased liver CB2 mRNA levels in 86% and diminished CB1 expression in 29% of patients. Finally, quality of life and Child-Pugh score improvements were reported in all patients [47]. This new piece of evidence demonstrated that PFD for two years treatment benefits CHC patients and improves inflammation, fibrosis and steatosis in higher number of patients as previously shown with 12-month treatment with PFD.
Role of PFD in the regulation of inflammation
This final section will attempt to describe the other side of PFD, the anti-inflammatory efficacy. The response to tissue injuries usually involves a large number of changes both local and distant from the site of damage. Inflammation takes place at the very beginning of the entire process to protect host, returning whether damaged is paled to normal functions. During inflammation, mainly circulating macrophages and neutrophils together with endothelial cells secrete TNF-α, IL-1β, and IL-6 pro-inflammatory cytokines. The order of secretion is important, since each cytokine fulfills a precise role in up-regulating or down-regulating the expression of others. These cytokines induce a number of local and systemic responses leading to more bloodstream inflammatory cell recruitment to the inflammation site. Sometimes the “tree must be cut to save the fruit”, this happens in the inflammatory process, which develops a violent response in trying to reduce the damage of harmful agent, but when it becomes chronic, alterations produced during tissue repair are often accomplished at the cost of “collateral damages”, such as large destruction of tissues. This is the reason why several physiological processes may also have anti-inflammatory activity. Therefore, the use of drugs that tackle at any point the inflammatory signaling pathway is of great importance for treatments aimed to reduce inflammation and its consequences.
There have been several studies in animal models of fibrosis where PFD immune modulation activities of have been evaluated. Macrophages are effector cells of innate response they are involved in the initiation and regulation of adaptive responses. Activated macrophages, can be divided into a M1 (inhibits cell proliferation and causes tissue damage exhibiting a Th1-like phenotype) and M2 macrophages (promotes cell proliferation and tissue repair exhibiting a Th2-like phenotype). Research has been conducted to analyse the effect of PFD in macrophages infiltration, where PFD has shown protective effects. Nephrectomised rats showed an increase in various molecules such as TNF-α, IL-6, and nitric synthase-2 oxide (expressed by macrophages M1), which decreased significantly their expression after PFD treatment. Also, a diminishment of arginase-1, dectin-1, CD206, and CD86 (expressed on macrophages M2) was observed. So it is proposed that, PFD efficacy is observed in both, early and late periods of fibrosis [52].
Other studies have shown that PFD alters T cell proliferation and cytokine release in response to T cell receptor (TCR) activation, regulating T cells (CD4+ CD25+) suppressive effects using an in vitro assay. Additionally, PFD effects on alloantigen-induced T-cell proliferation in vivo were assessed by adoptive transfer of carboxyfluorescein diacetate succinimidyl ester-labeled T cells across a parent->F1 major histocompatibility complex mismatch, as well as using a murine heterotopic cardiac allograft model (BALB/c->C57BL/6). PFD was found to inhibit the responder frequency of TCR-stimulated CD4+ cell total proliferation in vitro and in vivo, whereas both CD4 and CD8 proliferation index was reduced by PFD. Additionally, PFD inhibited TCR-induced production of multiple proinflammatory cytokines and chemokines [8]. Chen, JF et al. (2013), found that treatment with PFD prevents cytochrome C translocation from mitochondria and excluding in cytosol, resulting in the inhibition of caspase-9 and caspase-3 in kidney cortex of partial nephrectomised rats. PFD inhibits apoptosis by maintaining stability of the mitochondrial membrane, as observed in rat renal proximal tubular cells and human renal proximal tubular epithelial cell line (HK2), by inhibiting mitochondrial pathway of apoptosis signaling [53]. Also, acute injury becomes most evident at the time of nonspecific, chemokine-mediated amplification infiltrate recruitment of neutrophils [54]. PFD inhibited increases of inflammatory cytokines and cytokine-induced neutrophil chemoattractant (CINC) and iNOS in part by inhibition of NF-κB activation (Fig. 1) [45]. Therefore, among the main properties of PFD, reduction of inflammatory mediators is one of the most relevant.
Several questions relating PFD effects on the control of transcription factors and cytokine network remain unanswered. The knowledge of these complex processes and its regulatory pathway are essential in order to develop safe and effective anti-inflammatory therapies against pathologies where inflammation induced damaged is relevant. Currently, a phase II clinical trial is on-going in order to analyse PFD effects on Systemic Sclerosis-Related Interstitial Lung Disease (NCT01933334) [55]. Thus, this type of studies will provide solid evidence for a better understanding of mechanisms involved in the effects of anti-inflammatory and anti-fibrotic drugs.
Abbreviations
AF: atrial fibrillation; CB1: cannabinoid 1 receptor; CB2: cannabinoid 2 receptor; CHC: chronic hepatitis C; CINC: cytokine-induced neutrophil chemoattractant; eGFR: glomerular filtration rate; ESRD: end-stage renal disease; ET: Endothelin inhibitors I; FGF: fibroblast growth factor; FVC: forced vital capacity; HPS: Hermansky-Pudlak Syndrome; IPF: idiopathic pulmonary fibrosis; MMP-2: metalloproteinase 2; MMP-9: metalloproteinase 9; NAC: N-acetylcysteine; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; PDGF: platelet derived growth factor; PFD: pirfenidone; PFS: progression-free survival; TCR: T cell receptor; TGF-β: transforming grow factor β; TIMP-1: tissue inhibitor metalloproteinase; TNF-α: tumor necrosis factor α; α-SMA: α-smooth muscle Actin.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Huang NY, Ding L, Wang J, Zhang QY, Liu X, Lin HD, Hua WY. Pharmacokinetics, safety and tolerability of pirfenidone and its major metabolite after single and multiple oral doses in healthy Chinese subjects under fed conditions. Drug Res. 2013;63:388-95
2. Macias-Barragan J, Sandoval-Rodríguez AS, Navarro-Partida J, Armendariz-Borunda J. The multifaceted role of pirfenidone and its novel targets. Fibrogenesis Tissue Repair. 2010;3:16
3. Paz Z, Shoenfeld Y; Antifibrosis. To reverse the irreversible. Clinic Rev Allerg Immunol. 2010;38:276-86
4. Zamara E, Novo E, Parola M. Oxidative stress and liver fibrosis: from liver injury to the modulation of cell signaling and response. Ali S, Mann DA, Friedman SL, eds. Liver Diseases: Biochemical Mechanisms and New Therapeutic Insights Enfield, NH, USA: Science Publishers 2004:93-114
5. Salazar-Montes A, Ruiz-Corro L, López-Reyes A, Castrejón-Gómez E, Armendáriz-Borunda J. Potent antioxidant role of pirfenidone in experimental cirrhosis. Eur J Pharmacol. 2008;24:69-77
6. Seki E, Brenner DA. Recent advancement of molecular mechanisms of liver fibrosis. J Hepatobiliary Pancreat Sci. 2015
7. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028-1040
8. Visner GA, Liu F, Bizargity P, Liu H, Liu K, Yang J, Wang L, Hancock WW. Pirfenidone inhibits T cell activation, proliferation, cytokine and chemokine production, and host alloresponses. Transplantation. 2009;88:330-338
9. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788-824
10. Azuma A, Taguchi Y, Ogura T, Ebina M, Taniguchi H, Kondoh Y, Suga M, Takahashi H, Nakata K, Sato A, Kudoh S, Nukiwa T; Pirfenidone clinical study group in Japan. Exploratory analysis of a phase III trial of pirfenidone identifies a subpopulation of patients with idiopathic pulmonary fibrosis as benefiting from treatment. Respiratory Research. 2011;12:143
11. Cottin V. The role of pirfenidone in the treatment of idiopathic pulmonary fibrosis. Respiratory Research. 2013;14(Suppl 1):S5
12. Albera C, Ferrero C, Rindone E, Zanotto S, Rizza E. Where do we stand with IPF treatment? Respiratory Research. 2013;14(Suppl 1):S7
13. Hisatomi K, Mukae H, Sakamoto N, Ishimatsu Y, Kakugawa T, Hara S, Fujita H, Nakamichi S, Oku H, Urata Y, Kubota H, Nagata K, Kohno S. Pirfenidone inhibits TGF-β1-induced over-expression of collagen type I and heat shock protein 47 in A549 cells. BMC Pulmonary Medicine. 2012;12:24
14. Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167
15. Rafii R, Juarez MM, Albertson TE, Chan AL. A review of current and novel therapies for idiopathic pulmonary fibrosis. J Thorac Dis. 2013;5:48-73
16. Richeldi L, Yasothan U, Kirkpatrick P. Pirfenidone. Nature Reviews Drug Discovery. 2011;10:489-90
17. Castelino FV, Varga J. Interstitial lung disease in connective tissue diseases: evolving concepts of pathogenesis and management. Arthritis Res Ther. 2010;12:213
18. King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083-92
19. Taniguchi H, Kondoh Y, Ebina M, Azuma A, Ogura T, Taguchi Y, Suga M, Takahashi H, Nakata K, Sato A, Sugiyama Y, Kudoh S, Nukiwa T; Pirfenidone Clinical Study Group in Japan. The clinical significance of 5% change in vital capacity in patients with idiopathic pulmonary fibrosis: extended analysis of the pirfenidone trial. Respir Res. 2011;12:93
20. Loveman E, Copley VR, Colquitt J, Scott DA, Clegg A, Jones J, O'Reilly KM, Singh S, Bausewein C, Wells A. The clinical effectiveness and cost-effectiveness of treatments for idiopathic pulmonary fibrosis: a systematic review and economic evaluation. Health Technol Assess. 2015;19(20):1-336
21. 21. Kometani T, Okamoto T, Yoshida S, Yoshino I. Acute respiratory distress syndrome after pulmonary resection. Gen Thorac Cardiovasc Surg. 2013;61:504-12
22. Jiang C, Huang H, Liu J, Wang Y, Lu Z, Xu Z. Adverse events of pirfenidone for the treatment of pulmonary fibrosis: a meta-analysis of randomized controlled trials. PLoS One. 2012;7:47024
23. Onoue S, Seto Y, Kato M, Aoki Y, Kojo Y, Yamada S. Inhalable powder formulation of pirfenidone with reduced phototoxic risk for treatment of pulmonary fibrosis. Pharm Res. 2013;30:1586-1596
24. Couluris M. Treatment of idiopathic pulmonary fibrosis with losartan: a pilot project. Lung. 2012;190:523-527
25. O'Brien K, Troendle J, Gochuico BR, Markello TC, Salas J, Cardona H, Yao J, Bernardini I, Hess R, Gahl WA. Pirfenidone for the treatment of Hermansky-Pudlak Syndrome pulmonary fibrosis. Mol Genet Metab. 2011;103:128-134
26. Thielen N, Huizing M, Krabbe JG, White JG, Jansen TJ, Merle PA, Gahl WA, Zweegman S. Hermansky-Pudlak syndrome: the importance of molecular subtyping. J Thromb Haemost. 2010;8:1643-1645
27. Zamani N. Pirfenidone; can it be a new horizon for the treatment of pulmonary fibrosis in mustard gas-intoxicated patients? DARU Journal of Pharmaceutical Sciences. 2013;21:13
28. Gan Y, Herzog EL, Gomer RH. Pirfenidone treatment of idiopathic pulmonary fibrosis. Therapeutics and Clinical Risk Management 2011. 2014;7:39-47
29. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-82
30. FDA.gov: Silver Spring (MD). FDA News Release: FDA approves Esbriet to treat idiopathic pulmonary fibrosis. Revised 24 Mar 2015. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm418991.htm
31. Cho ME, Kopp JB. Pirfenidone: an anti-fibrotic and cytoprotective agent as therapy for progressive kidney disease. Expert Opin Investig Drugs. 2010;19:275-283
32. Tanios BY, Ziyadeh FN. Emerging therapies for diabetic nephropathy patients: beyond blockade of the renin-angiotensin system. Nephron Extra. 2012;2:278-282
33. Sharma K, Ix JH, Mathew AV, Cho M, Pflueger A, Dunn SR. Pirfenidone for diabetic nephropathy. Am Soc Nephrol. 2011;22:1144-1151
34. Anil Karihaloo. Anti-fibrosis therapy and diabetic nephropathy. Curr Diab Rep. 2012;12:414-422
35. Shi Q, Liu X, Bai Y, Cui C, Li J, Li Y, Hu S, Wei Y. In vitro effects of pirfenidone on cardiac fibroblasts: proliferation, myofibroblast differentiation, migration and cytokine secretion. PLoS One. 2011;6:28134
36. Tamargo J, López-Sendón J. Novel therapeutic targets for the treatment of heart failure. Nature Reviews Drug Discovery. 2011;10:536-555
37. Wang Y, Hou X, Li Y. Association between transforming growth factor β1 polymorphisms and atrial fibrillation in essential hypertensive subjects. Journal of Biomedical Science. 2010;17:23
38. Yamazaki T, Yamashita N, Izumi Y, Nakamura Y, Shiota M, Hanatani A, Shimada K, Muro T, Iwao H, Yoshiyama M. The antifibrotic agent pirfenidone inhibits angiotensin II-induced cardiac hypertrophy in mice. Hypertens Res. 2012;35:34-40
39. ClinicalTrials.gov. Pirfenidone to Treat Hypertrophic Cardiomyopathy. http://clinicaltrials.gov/ct2/show/NCT00011076?term=NCT00011076&rank=1
40. ClinicalTrials.gov. Heart Function and Exercise Capacity in Patients With Hypertrophic Cardiomyopathy. http://clinicaltrials.gov/ct2/show/NCT00354328
41. Zhong H, Sun G, Lin X, Wu K, Yu M. Evaluation of pirfenidone as a new postoperative antiscarring agent in experimental glaucoma surgery. Invest Ophthalmol Vis Sci. 2011;52:3136-42
42. Sun G, Lin X, Zhong H, Yang Y, Qiu X, Ye C, Wu K, Yu M. Pharmacokinetics of pirfenidone after topical administration in rabbit eye. Mol Vis. 2011;17:2191-6
43. Choi K, Lee K, Seung-Wook R, Im M, Kook KH, Choi C. Pirfenidone inhibits transforming growth factor-β1-induced fibrogenesis by blocking nuclear translocation of Smads in human retinal pigment epithelial cell line ARPE-19. Mol Vis. 2012;18:1010-1020
44. Yang Y, Ye Y, Lin X, Wu K, Yu M. Inhibition of pirfenidone on TGF-beta2 induced proliferation, migration and epithlial-mesenchymal transition of human lens epithelial cells line SRA01/04. PLoS One. 2013;8:56837
45. Kim H, Choi YH, Park SJ, Lee SY, Kim SJ, Jou I, Kook KH. Antifibrotic effect of pirfenidone on orbital fibroblasts of patients with thyroid-associated ophthalmopathy by decreasing TIMP-1 and collagen levels. Invest Ophthalmol Vis Sci. 2010;51:3061-6
46. Armendáriz-Borunda J, Islas-Carbajal MC, Meza-García E, Rincón AR, Lucano S, Sandoval AS, Salazar A, Berumen J, Alvarez A, Covarrubias A, Aréchiga G, García L. A pilot study in patients with established advanced liver fibrosis using pirfenidone. Gut. 2006;55:1663-1665
47. Flores-Contreras L, Sandoval-Rodríguez AS, Mena-Enriquez MG, Lucano-Landeros S, Arellano-Olivera I, Alvarez-Álvarez A, Sanchez-Parada MG, Armendáriz-Borunda J. Treatment with pirfenidone for two years decreases fibrosis, cytokine levels and enhances CB2 gene expression in patients with chronic hepatitis C. BMC Gastroenterol. 2014;14:131
48. Tsuchiya H, Kaibori M, Yanagida H, Yokoigawa N, Kwon AH, Okumura T, Kamiyama Y. Pirfenidone prevents endotoxin-induced liver injury after partial hepatectomy in rats. J Hepatol. 2004;40:94-101
49. Wang F, Wen T, Chen XY, Wu H. Protective effects of pirfenidone on D-galactosamine and lipopolysaccharide-induced acute hepatotoxicity in rats. Inflamm Res. 2008;57:183-188
50. García L, Hernández I, Sandoval A, Salazar A, Garcia J, Vera J, Grijalva G, Muriel P, Margolin S, Armendariz-Borunda J. Pirfenidone effectively reverses experimental liver fibrosis. J Hepatol. 2002;37:797-805
51. Di Sario A, Bendia E, Macarri G, Candelaresi C, Taffetani S, Marzioni M, Omenetti A, De Minicis S, Trozzi L, Benedetti A. The anti-fibrotic effects of pirfenidone in rat liver fibrosis is mediated by downregulation of procollagen alpha 1(l), TIMP-1 and MMP2. DigLiverDis. 2004;36:744-751
52. Chen JF, Ni HF, Pan MM, Liu H, Xu M, Zhang MH, Liu BC. Pirfenidone inhibits macrophage infiltration in 5/6 nephrectomized rats. Am J Physiol Renal Physiol. 2013;15:304
53. Chen JF, Liu H, Ni HF, Lv LL, Zhang MH, Zhang AH, Tang RN, Chen PS, Liu BC. Improved mitochondrial function underlies the protective effect of pirfenidone against tubulointerstitial fibrosis in 5/6 nephrectomized rats. PLoS One. 2013;8:83593
54. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43(2 Suppl 1):S54-62
55. ClinicalTrials.gov: Bethesda (MD). Safety and tolerability of pirfenidone in patients with systemic sclerosis-related interstitial lung disease (SSc-ILD) (LOTUSS). Identifier: NCT01933334. Revised 2 Oct 2014. http://clinicaltrials.gov/ct2/show/study/NCT01933334
Author contact
![]() Corresponding author: armdbocom
Corresponding author: armdbocom