Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(2):163-176. doi:10.7150/ijms.10826 This issue Cite
Research Paper
Identification of Novel Compounds against an R294K Substitution of Influenza A (H7N9) Virus Using Ensemble Based Drug Virtual Screening
Nhut Tran1,2, Thanh Van1,2, Hieu Nguyen1, Ly Le1,2, ![]()
1. Life Science Laboratory, Institute for Computational Science and Technology at Ho Chi Minh City, SBI building, Quang Trung Software City, Tan Chanh Hiep Ward, District 12, Ho Chi Minh City, Vietnam
2. School of Biotechnology, International University - Vietnam National University Ho Chi Minh City, Quarter 6, Linh Trung Ward, Thu Duc District, Ho Chi Minh City, Vietnam
Received 2014-10-16; Accepted 2014-12-16; Published 2015-1-12
Abstract
Influenza virus H7N9 foremost emerged in China in 2013 and killed hundreds of people in Asia since they possessed all mutations that enable them to resist to all existing influenza drugs, resulting in high mortality to human. In the effort to identify novel inhibitors combat resistant strains of influenza virus H7N9; we performed virtual screening targeting the Neuraminidase (NA) protein against natural compounds of traditional Chinese medicine database (TCM) and ZINC natural products. Compounds expressed high binding affinity to the target protein was then evaluated for molecular properties to determine drug-like molecules. 4 compounds showed their binding energy less than -11Kcal/mol were selected for molecular dynamics (MD) simulation to capture intermolecular interactions of ligand-protein complexes. The molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) method was utilized to estimate binding free energy of the complex. In term of stability, NA-7181 (IUPAC namely {9-Hydroxy-10-[3-(trifluoromrthyl) cyclohexyl]-4.8-diazatricyclo [6.4.0.02,6]dodec-4-yl}(perhydro-1H-inden-5-yl)formaldehyde) achieved stable conformation after 20ns and 27ns for ligand and protein root mean square deviation, respectively. In term of binding free energy, 7181 gave the negative value of -30.031 (KJ/mol) indicating the compound obtained a favourable state in the active site of the protein.
Keywords: Influenza virus, neuraminidase, virtual screening, molecular dynamic simulation, binding free energy
Introduction
The emergence of new subtypes of influenza A virus in recent years has alarmed us a highly pathogenic change in influenza A virus subtypes [1-3]. Among which, H7N9, a new subtype of influenza A virus (designated A/Shanghai/2/2013, and A/Anhui/1/2013), caused an epidemic in China, and killed 46 patients among 144 infectious cases in 2013. In 2014, influenza A H7N9 virus continuously spreads throughout the population. As of February 2014, there are a total of 375 cases including 115 deaths reported in China, Taipei, Hong Kong and Malaysia [4]. Although Hemagglutinin (H7) of influenza A virus regularly circulates among birds, its infection to human was rarely found. The combination of H7 and N9 was only found in birds and some outbreaks were reported in the Netherlands, Japan, and the United States [5, 6]. There are no human infectious cases with H7N9 virus found until 2013, when the first infected case in human with neuraminidase serotype N9 of H7N9 subtype was reported in March, in Shanghai, China [7].
Despite the fact that annual vaccination is the most effective ways to reduce the rate of infections and deaths relating to influenza virus [8], vaccine is unlikely to be effective in response to newly emerging influenza virus due to unavailability of new specific vaccines [9, 10]. Hence, there is a high risk in fighting against a new influenza pandemic while people are waiting for approvable vaccine supplies. In order to eliminate this risk, scientists put their immense effort to develop new effective antiviral drugs [11, 12]. Two different classes of influenza virus inhibitors targeting M2 channel protein and neuraminidase that are approved by the FDA available for treatment of influenza virus infections include; adamantane drugs (amantadine and rimantadine) [13] and neuraminidase (NA) inhibitors (Zanamivir and Oseltamivir) [14]. The adamantine was administered to treat influenza A virus while NA inhibitors has been approved for both influenza A and influenza B viruses.
Nevertheless, influenza viruses have become resistance to existing influenza anti-viral drugs, which is more deadly to human and increases the probability of emerging the next influenza pandemic. By the end of 2009-2010, all H1N1 pandemic virus tested samples showed resistance to both Amatadine and Rimatadine, two adamantane-based drugs targeting M2 protein channel; the resistance is blamed by single mutation in the M2 protein channel [15-17]. The most remarkable mutation is in S31N position that maintains the function of M2 channel in the present of adamantine drugs [18-21]. In addition, the loss of ability to bind and inhibit the function of NA in influenza viruses results in NA inhibitor resistance. Single amino acid change known as the “H275Y” mutation in 2009 H1N1 flu virus [22-24] and “R292K” mutation in influenza A virus [25-30] is conferred by drug resistance. Orientation and stabilization of various inhibitors in NA protein depend major on two or three Arginine residues in 150 loop [31], R292K mutation succeeded to unbalance stability and orientation of these inhibitors leading to drug resistance. Unexpectedly, all novel influenza A H7N9 viruses possess the mutation S31N in M2 protein channel and N9 serotype (designated as A/Shanghai/1/2013) encoded for R294K mutation [32-34].
Influenza virus NA plays a vital role in virus replication by cleaving the linkage between Sialic acid groups of the virus and glycoproteins locating on the surface membrane of the host cells, it also facilitates the spread of progeny virions infecting to a new host cell [35, 36]. Regarding the mutation resistance to existing NA inhibitors, NA protein has become a crucial target for drug design and development in controlling disease progression [11, 37]. In our study, molecular docking and dynamic approach have been employed to identify potential compounds that effectively inhibit the function of a newly resistant strain of NA. Virtual screening using ensemble based method was applied to screen molecules retrieved from the TCM database and ZINC natural products.
Materials and Methods
Protein preparation
The coordinations of H7N9 NA in inhibitor resistant and non-resistant strain were retrieved from the Protein Data Bank with PDB entries 4MX0 and 4MWV, respectively. Peramivir [38], Oseltamivir [39], Zanamivir [40], Laninamivir [41] and Sialic acid were obtained from Drug bank and served as control for rigid docking experiment. For virtual screening TCM database, the only resistant strain (4MX0) was used to generate a set of conformation for later ensemble based docking; molecular dynamic simulation was performed on the mutant using the program GROMACS (version 4.5.5) with the calculation method (force field) GROMOS96 53A6 [42-44], It was indicated that the new parameters set in force field GROMOS96 53A6 perform for protein as well as parameters set in force field GROMOS 45A3 and DNA in force field GROMOS 45A4, and it was found better performing in the folding-unfolding balance of the peptide [45]. The study of comparing 10 different force fields also confirmed that GROMOS 53A6 emerged to be one of two best force field for protein and peptide simulation [46]. The crystal structure of NA (4MX0) serves as the initial conformation. SPC water molecules were used to solvate the protein and counter ions were added to neutralized the total charge of the system[47]. A 154 μM NaCl was bathed into the system to mimic the salt concentration of human physiological condition. The solvated system was then energy-minimized by 5000 steps. The simulations were run under periodic conditions with NVT ensemble by using V-rescale coupling algorithm for maintaining temperature at 300K for 100-ps and NPT ensemble by using Nose Hoover coupling algorithm to constant the pressure at 1bar for 100-ps. The MD production was run on NPT condition at 300K for 50-ns. The time step for simulation was 2-fs. The PME algorithm[48] with 1.2 nm cut-off for real-space calculation was used to calculated the electrostatic interactions; a cut-off of 1.2 nm was used for van der Waals interactions.
Extract Representative MD Structures using RMSD Clustering
The MD simulation creates a huge of trajectory and in the effort to reduce assets of conformation for ensemble basesd docking; root mean square deviation (RMSD) clustering was employed to generate a set of representative structures with the aid of linkage method in GROMAC package. In this method, MD trajectories were gathered into different groups based on the RMSD and the average of each cluster was considered as a representative structure. Briefly, simulation frames were extracted from the MD simulation at every 2 ps over the 20 ns of the stable interval. The result of 2×103 trajectory structures was obtained and then superimposed using all Cα atoms to eliminate significant rotation and translation of the whole system. The clustering was performed based on framework and binding site residues including 117-119, 133-138, 146-152, 156, 179, 180,196-200, 223-228, 243-247, 277, 278, 293, 295, 344-347, 368,401, 402, and 426-441 (numbering in H5N1) (Figure 1 ) [39], these residues served as reference for clustering MD trajectory structures with a cut-off range 0.5 - 1 Å. After comparing the number of cluster populations to the total number of clusters in the simulation, a cut-off of 0.90 Å was selected. For each cluster, an average conformation was selected as a cluster representative structure, 52 structures were found in the clustering. With population sizes less than 100 conformations, these clusters were rejected. Hence, 25 structures with population sizes greater than 100 achieved 89.6% were served as receptors for docking.
Multiple sequence alignment of three different NA from H5N1, H7N9 Shanghai and H7N9 Anhui virus with PDB code 2HTY, 4MX0 and 4MWV, respectively. Numbering sequence based on H5N1 with rectangle indicating framework and binding site residues.

Compound library retrieval and drug-like molecule filter
Traditional medicine has been applied for thousand years to cure the disease in China and Asia region; it still plays an essential role in medical field today. Traditional Chinese medicine (TCM) database is the largest traditional medicine database in the world, which provides more than 20,000 pure compounds for drug screening in silicon [49]. In this study, we aimed to find the potential compounds in inhibiting NA by screening virtual library of Traditional Chinese medicine database (TCMD) as well as ZINC natural products. The TCMD library was downloaded from website http://tcm.cmu.edu.tw/. These complex files were separated into single pdb file and then all compounds with molecular weight greater than 500 Dalton were discharged.
Structure based virtual screening by ensemble based docking
Virtual screening is an effective method that has been widely utilized to filter a huge database of small organic molecules against a specific target protein [50, 51]. It enables us to identify potential compounds that highly chance to be drug candidates among thousand compounds. In this docking experiment, a set of 25 representative structures were utilized as receptors for ensemble based docking against selected compounds filtered from TCMD. Autodock Tools version 1.5.6rc3[52] was employed to merge polar hydrogen atoms, assign Gasteiger charges [53] to each of these structures. The grid box of each conformation was created to cover all binding site residues. The binding boxes were designed with the volume of 30 x 36 x 30 Å and 1 Å spacing.
The compounds with molecular weight less than 500 Dalton selected from TCM and ZINC natural products were included in the screening process. These compounds were then optimized geometrically to eliminate all overlapping and improper bonding by MOPAC 2012 package using the PM7 Hamiltonian Restricted Hartree-Fock method. Autodock Tools version 1.5.6rc3 [54, 55] was also used to merge polar hydrogen atoms add gangster charge and assign rotatable bond. This software was also used to add polar hydrogen atoms and assign gangster charge to representative structures of NA. The AutoDock vina performed the docking simulation using Lamarckian genetic algorithm [56]. During the docking procedure, ligands were flexible whereas the receptor was fixed. Docking parameters were set as a default with exhaustiveness of 400. Binding affinity from docking results was sorted in descending using homemade python script. All of the top hits with the binding affinity greater than 9 were chosen for further analysis.
ADME properties
The most effective and convenient way to deliver drugs to the circulatory system is through oral route, thus membrane permeability, bioactivity and toxicity are a vital consideration for drug development. As a pioneer, Linsperki is the first success to build a rule for filtering drug-like and non drug-like compounds [57]. The rule claims that successful candidates for drug development should conform basic molecular properties comprising Molecular Weight ≤500, calculated octanol-water partition coefficient logP ≤5, hydrogen bond donors ≤5 and acceptors ≤10. In our study, Lipinski's Rule of Five was applied to test the bioavailability characteristics of the top hit compounds. The molecular properties of top hits compounds were calculated online by using MOLINSPIRATION web server (http://www.molinspiration.com/).
MD simulation of the NA in complex with the four best docking compounds
Simulation was conducted using the program GROMACS (version 5.4.5) with the calculation method (force field) GROMOS[42, 43]. Docked poses from previous molecular docking jobs were used as a starting conformation for the molecular dynamics calculation. The water molecule Single Point Charge (SPC) was then added to the system[47]. Meanwhile, the ions (counter ions) were randomly placed to neutralize the simulation system with the concentration of 154 μM NaCl. The whole system was optimized energy with 5000 steps to ensure the system in which the atomic distance was not too close or the improper geometry did not match up each other. Then the whole system was run in the NVT periodic boundary conditions using the V-rescale at 300K for 100-ps and NPT next run at the Hoover algorithm at 1bar pressure for 100-ps. Molecular simulation production was run under NPT conditions at 300K for 50-ns to find stable conformations of each protein structure. The simulation time step was 2-fs. The electrostatic interactions were calculated using the PME algorithm[48] with a 1.2 nm cut to calculate the real space; a cut-off 1.2 nm-used van der Waals interactions
Binding free energy calculation
Although ensemble based docking was considered receptor as partial flexibility through docking many different structures, a correct prediction of the binding affinity is still missing. In our study, the correct binding free energy was calculated using MM/PBSA method. The method improves our docking energy by included protein flexibility and gives detailed energy composition. However, MM/PBSA method treats water implicitly thus, we might miss some explicit water-drug interactions which are important for drug binding but the method enable us calculate faster than implicitly method which require much more computer resources. Although the entropy contribution was excluded in our calculations, they do not significant affect to our results since we use the same protein for potential compounds screening, hence entropy contribution were assumed to be the same for all systems. The g_mmpbsa [58] was employed for calculating the component energy of the system and the best four compounds that had binding affinity greater than -11Kcal/mol were selected for binding free energy calculation. Particularly, the binding free energy of ligand-protein complex in solvent was expresses as:
∆Gbinding = Gcomplex - (Gprotein + Gligand)
where Gcomplex is the total free energy of the protein-ligand complex, Gprotein and Gligand are total energy of separated protein and ligand in solvent, respectively. The free energy for each individual Gcomplex ,Gprotein and Gligand were estimated by:
Gx = ‹EMM› + ‹Gsolvation›
where x is the protein, ligand, or complex. EMM is the average molecular mechanics potential energy in vacuum and Gsolvation is free energy of solvation. The molecular mechanics potential energy was calculated in vacuum as following:
EMM = Ebonded + Enon-bonded = Ebonded + (Evdw + Eelec)
where Ebonded is bonded interaction including of bond, angle, dihedral and improper interactions and Enon-bonded is non-bonded interactions consist of van der Waals (Evdw ) electrostatic (Eelec) interactions.
The solvation free energy (Gsolvation) was estimated as the sum of electrostatic solvation free energy (Gpolar) and apolar solvation free energy (Gnon-polar) :
Gsolvation = Gpolar + Gnon-polar
where Gpolar was computed using the Poisson-Boltzmann (PB) equation [59] and Gnon-polar estimated from the solvent-accessible surface area (SASA) as equation following:
Gnon-polar = γSASA + b
where γ is a coefficient related to surface tension of the solvent and b is fitting parameter.
Results
Comparison of biding affinity of the crystal structure of Anhui and Shanghai virus to defined inhibitors
The crystal structures of NA in Shanghai and Anhui virus were selected for docking with Oseltamivir, Zanamivir, Peramivir, Laninarmivir and Sialic acid. Remarkably, all inhibitors in complex with NA of Shanghai virus (4MX0) showed their binding affinity lower than those of NA of Anhui virus (4MWV) (Table 1). Particularly, compared to Anhui virus NA, the complex of Shanghai virus NA with Oseltamivir showed the decrease of 0.5 Kcal/mol while its complex with Peramivir dropped down 0.7 Kcal/mol. This falling was repeated in Zanamivir and Laninarmivir, but it was relatively small, with 0.3 Kcal/mol in Zanamivir, and 0.6 Kcal/mol in Laninarmivir. The docking results of H7N9 NA agreed well with the experiential results in which NA R292K substitution was highly resistant to Oseltamivir and Peramivir and partially resistant to Zanamivir [33]. Amazingly, the substrate (Sialic acid) unchanged their binding affinity (-7.0 Kcal/mol) that was greater than Oseltamivir and Laninarmivir, equal to Zanamivir and less than Peramivir. As a result of competitive inhibition, the Sialic acid strongly competed for the binding site of NA since it has lower binding affinity than Oseltamivir and Laninarmivir. In complex with Zanamivir, both the substrate and the inhibitor have the same binding affinity to the NA. Hence they both had a chance to interact with NA. These results explain experimental data that R294K substitution led to extreme resistance of NA to Oseltamivir and conferred less resistance to Peramivir, Zanamivir and Laninarmivir [29, 30].
Binding affinity of NA N9 with four different inhibitors and a substrate
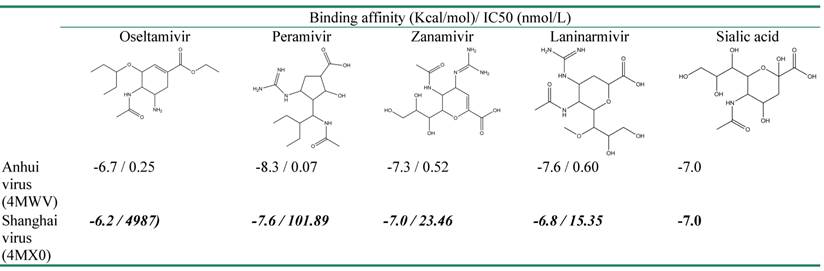
Experimental data of IC50 used for comparison with binding affinity was taken from the work of Katrina Sleeman, Zhu Guo, et al, 2013.
Comparison of molecular interaction of the crystal structure of Anhui and Shanghai virus to defined inhibitors
R294 is a highly conserved residue across all NA subtypes, and it, together with two other highly conserved residues (R119 and R372), forms an arginine triad in the enzyme active size [60]. R294K substitution has rarely occurred and to date has only been reported from the patients treated with Oseltamivir [60,61]. Recently, influenza H7N9 (A/Shanghai/1/2013) has become the latest strain possessing this mutation. To understand the interaction in detail, hydrogen bond and hydrophobic interaction were analyzed (Table 2). The parameters for hydrogen bond detection were set with 3Å of Hydrogen-Acceptor distance cut-off, 2.25Å of Donor-Acc distance cut-off, sp2, sp3 donor- hydrogen-acceptor angle range 1200 - 1800 and sp2, sp3 donor-acceptor-acceptor N angle range 1100 - 1500. In Anhui virus, the docking results indicated that Sialic acid and all inhibitors except Oseltamivir formed a hydrogen bond with NA at R119. Moreover, a hydrogen bond forming was observed between R294 residue with all inhibitors and the substrate except Zanamivir. R372 residue is considered as the most important site for drug binding when it formed a hydrogen bond to all inhibitors and the substrate. In Shanghai virus, there was a significant reduce in the number of hydrogen bonds to all inhibitors which made NA less sensitive to the drugs. In contrast, Sialic acid relatively remained the number of hydrogen bonds to NA. In particular, the four most important residues comprising R119, R294, R372 and R153 remained hydrogen bonding to Sialic acid, and there was only one hydrogen bond of residue D152 shift to residue E120. This explains the conservation in binding affinity between wild type and mutant of NA to the substrate. In the other hand, these hydrogen bonds were entirely absent in Oseltamivir, Zanamivir and Laninarmivir while only Peramivir remained hydrogen bonds with R119, R372 and an alternative bonding with W180. Regarding binding affinity, the fall in the number of hydrogen bonds of inhibitors leads to decrease binding affinity despite the increase in hydrophobic interaction residues.
Hydrogen bonds and hydrophobic interaction residue to inhibitors and substrate.
| Anhui Virus (4MWV) | Shanghai virus (4MX0) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| O | P | Z | L | S | O | P | Z | L | S | |
| HB interaction residues | R119 | R119, | R119 | R119 | R119 | R119 | ||||
| E120 | E120 | |||||||||
| D152 | ||||||||||
| R153 | R153 | R153 | R153 | |||||||
| W180 | W180 | |||||||||
| R294 | R294* | R294 | R294 | K294 | ||||||
| R372 | R372* | R372 | R372 | R372* | R372* | R372 | ||||
| Y406 | Y406 | Y406 | ||||||||
| HP & Elec interaction residues | R119 | R119 | R119 | R119 | ||||||
| E120 | E120 | E120 | E120 | E120 | E120 | |||||
| D152 | D152 | D152 | D152 | D152 | D152 | D152 | ||||
| R153 | R153 | R153 | ||||||||
| R157 | R157 | R157 | R157 | |||||||
| W180 | W180 | W180 | W180 | W180 | ||||||
| S181 | ||||||||||
| I224 | ||||||||||
| R226 | R226 | R226 | R226 | R226 | R226 | |||||
| E229 | E229 | E229 | E229 | E229 | ||||||
| E278 | E278 | E278 | E278 | E278 | ||||||
| E279 | E279 | E279 | E279 | |||||||
| R294 | K294 | K294 | K294 | K294 | ||||||
| N296 | ||||||||||
| R372 | R372 | R372 | ||||||||
| Y406 | Y406 | Y406 | Y406 | Y406 | Y406 | |||||
HB: Hydrogen bond, HP: Hydrophobic. Elec: Electrostatic. (*): 2 hydrogen bonds O: Oseltamivir,
P: Peramivir, Z: Zanamivir, L: Laninarmivir, S: Sialic acid
Stability and clustering of NA of simulation system
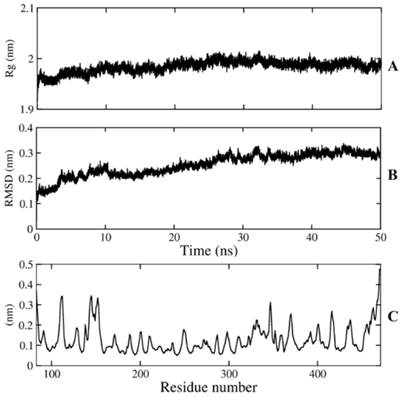
The MD simulation was performed to guarantee the stability of the model over 50ns of simulation time. The protein got the stable coordination after 30 ns with an average backbone root mean square deviation (RMSD) of ~ 0.28 Å (Figure 2). The size and the shape of NA maintained compact during 50ns simulation time by analyzing radius of gyration with gyration radii (Rg) of ~ 1.98 Å. The fluctuations of individual residues were also investigated to determine the mobility of every residue over simulations, root mean square fluctuations (RMSF) of Cα atoms indicated that both N and C terminal reached the maximum fluctuation with ~ 3.6Å and ~ 4.8Å, respectively, while most of framework and binding site residues fell into a range of 1-2Å. The stability of framework and binding site residues indicated the importance of these residues in conservation of NA function.
Conformational analysis of NA. A) radius of gyration, B) Cα RMSD (Rg), C) Cα RMSF.
Virtual screening and bioactivity analysis
Virtual screening helps us to identify potential inhibitors for a target protein; the top hit identification is collected with the aid of docking engine through huge virtual structure libraries on a target protein. This application facilitates us to cut down time and effort expenses by selecting compounds which have high bioactivity for experimental session and increase successful probability in vitro experiments. In the effort to identify the new lead compounds from TMCD, we performed assemble-based docking against selective compounds in the database to identify the top hit for a new strain of NA. 24 compounds in database showed their binding affinity greater than -9 Kcal/mol were obtained and served as top hits for further analysis (Table 3). The new lead compounds need properties that would make them become more likely to penetrate through membrane as well as easy to be absorbed by human body. The molecular properties shown in Table 3 let us select chemical compounds having a pharmacological or biological activity that can make them an orally active drug in human. It was observed that all top hit compounds possessed a high lipophilicity which maintained the penetration of compounds through cell membrane; however, the number of hydrogen bond donors is less than that of the control which indicated that the solubility was less than that of the inhibitors.
Binding affinity and molecular properties of top compounds.


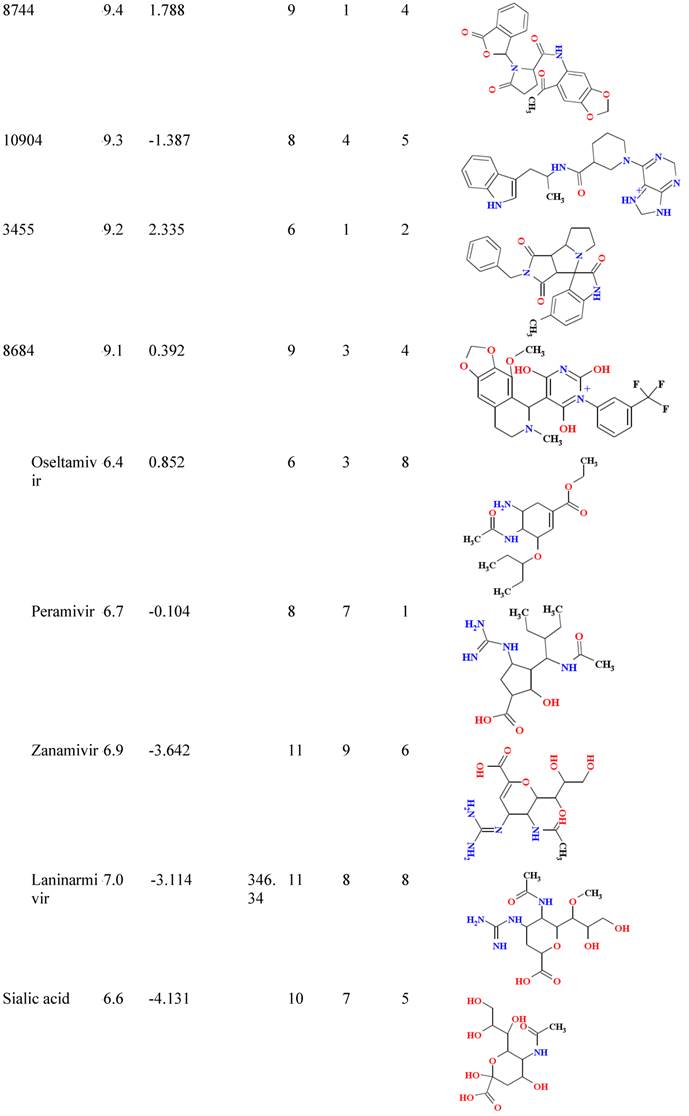
B/A: Binding affinity; MW: molecular weight; Hb-A: hydrogen bond acceptor, Hb-D hydrogen bond donor; Nrtb: number of rotatable bond
MD simulations of the complex
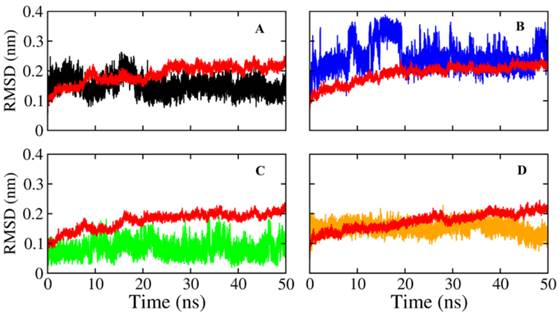
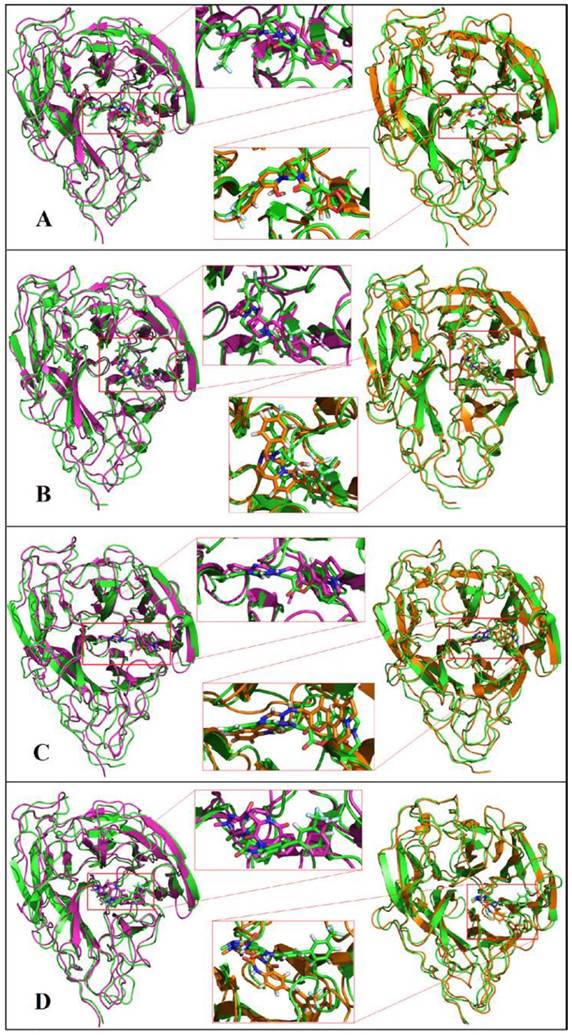
The docking result provides only a hard view of ligand-receptor interactions since its receptor were kept rigid or partially flexible in ensemble based and flexible docking. Molecular dynamic simulation helps us fulfill gaps in docking experiment that did not include protein flexibility and movement relating to the stability of the complex interaction. In this study, we also performed molecular dynamic simulation on receptor-ligand complex in order to figure out the interactions in free movement of the complex system, changing in residues as well as dependent and independent movement of the protein-ligand complex. Top 4 ligands with binding affinity in docking results less than -11 Kcal/mol were included for MD simulation. The RMSD for backbone of NA in all system through 50 ns is shown in Figure 3, the average movement of protein atoms were less than 0.7 Å in three system comprising NA in complex with 10877, 7182 and 7181 after 27 ns of simulation. This means the protein achieved its stable conformation at the time 27ns and kept that conformation through the rest of simulation time with an average backbone RMSD of ~0.2 to ~ 0.21 Å. In consideration of NA-40 complex, the backbone RMSD of NA did not reach its stable coordination after 27ns simulation as other system and continuously increased until the end of 50ns simulation. Analyzing RMSD of separate ligands also indicated that the movement as well as rotation of 7181 and 10877 were not significant compared to the original position while 7182 strongly change in position at two period intervals; from 15 to 20 ns and from 47 to 50 ns. In the last case, although ligand 40 remains original position through 37ns, it is strong rotation and movement away from docking position. Moreover, it was observed from Figure 4 that the ligands tend to move toward substrate binding site. In particular, the polar tail of three out of four compounds move toward active site residues and two of them (7181, 10877) remain that location till the end of simulation.
Root mean square deviations of proteins and ligands. The backbone RMSD of NA of Shanghai virus was colored in black while its corresponding ligand was colored in red. A) RMSD of NA and its corresponding ligand 7181; B) RMSD of NA and its corresponding ligand 7182; C) RMSD of NA and its corresponding ligand 40; D) RMSD of NA and its corresponding ligand 10877.
Molecular dynamics simulations of NA in complex with ligands A) 7181, B) 7182, C) 10877, D) 40. Initial coordination was displayed in pink; 30ns simulation was displayed in green and 50ns simulation in orange.
Binding free energy analysis
The best four ligands in complex with NA from molecular dynamic simulation were used to calculate binding free energy using MM-PBSA method. Snapshots were extracted at every 16-ps of stable intervals from 20ns MD trajectory and served as input for calculation. The binding free energy and its corresponding components obtained from the MM/PBSA calculation of the NA-ligand complexes were listed in Table 4. The results indicated that two out of four compounds possessed a negative binding free energy with -30.031 and -36.553 for 7181 and 7182, respectively. Moreover, van der Waals, electrostatic interactions and non-polar solvation energy negatively contribute to the total interaction energy while only polar solvation energy positively contributes to total free binding energy. In term of negative contribution, van der Waals interaction give much larger than electrostatic interactions for all cases except for NA-40 complexes in which van der Walls interaction contribute significantly lower than electrostatic interaction. The non-polar free energy contributes relatively small compared to total energy. This indicates that non-polar solvation energy, van der Waals and electrostatic interaction together contribute to the complex stability. In spire polar solvation energy was the only positive one; its value was as large as the sum of all other components. Therefore, the total free binding energy depends much more on this value.
Free energy and their components (KJ/mol) of the NA in complex with ligands.
| Ligand ID | Eelec | Evdw | Gsol -polar | Gsol-non-polar | ∆Gbinding |
|---|---|---|---|---|---|
| 7181 | -196.351 ±12.9 | -291.432 ±32.2 | 478.350 ±48.5 | -20.598 ±1.2 | -30.031 ±28.8 |
| 7182 | -195.696 ±0.4 | -244.988 ±1.4 | 424.647 ±1.8 | -20.547 ±0 | -36.553 ±1 |
| 10877 | -162.212 ±14.2 | -401696 ±47.3 | 608.787 ±63.6 | -17.936 ±1.6 | 26.942 ±28.4 |
| 40 | -142.347 ±39.4 | -88.305 ±29.2 | 274.847 ±76.8 | 16.524 ±4.4 | 27.671 ± 32.5 |
Conclusion
The emergence of new influenza virus subtype H7N9 in recent years has been causing a big trouble to global heath since it is high virulence and mortality, and Shanghai strains have already acquires resistance to existing anti-viral drugs for influenza treatment. In this study, we demonstrated resistant mechanism of mutant strain H7N9 (R294K) in Shanghai virus by comparing binding poses and binding affinity between inhibitors as well as the substrate and X-ray structure of mutant and wild type NA in H7N9 virus. Moreover, we also performed virtual screening of more than 57,000 compounds retrieved from traditional Chinese medicine (TCM) database and ZINC natural products against NA of Shanghai virus using ensemble based docking approach. 24 compounds expressed the binding affinity greater than -9 kcal/mol, 4 compounds among these 24 top hits showed their binding affinity greater than -11kcal/mol which is an indicator of drug candidate for drug development. In addition, the molecular dynamic simulation and binding free energy analysis was took in account to validate and capture intermolecular interaction through time evolution. 3 of 4 best potential compounds have been observed getting the stable conformation after 27-ns of simulation time and 2 of them expressed negative value in binding free energy. Therefore, we concluded that 7181 is the best potential compound for NA (R294K) inhibition and should be further analyzed and served as drug candidate for in vivo testing. The limitation of our work comes from binding free energy calculation. We calculated relative binding free energy without entropy energy contribution. Although entropy energy should be considered in order to obtain absolute binding free energy, we use only one target protein for screening potential compounds and we assume entropy energy contribution of the complex with similar protein was not significantly difference. Moreover, in term of binding free energy individual residues energy needs to be analyzed to examine important residues that contribute to system stability.
Acknowledgements
The work was funded by the Department of Science and Technology at Ho Chi Minh City under grant number 121/TB-SKHCN. Computing resources provided by the Institute for Computational Science and Technology—Ho Chi Minh City is gracefully acknowledged.
Competing Interests
The authors declare that the research work in this manuscript contain no material previously published or written by another person except where due references are made. There are no affiliations with or involvement in any organization or entity with any financial interest, or non-financial interest in the subject matter or materials discussed in this manuscript.
References
1. Kilbourne ED. Influenza pandemics of the 20th century. Emerging infectious diseases. 2006;12:9-14 doi:10.3201/eid1201.051254
2. Hafner S. Birds of ill omen--is H7N9 the harbinger of the next pandemic? Microbes and infection / Institut Pasteur. 2013;15:429-31 doi:10.1016/j.micinf.2013.04.011
3. Schenk C, Plachouras D, Danielsson N, Nicoll A, Robesyn E, Coulombier D. Outbreak with a novel avian influenza A(H7N9) virus in China--scenarios and triggers for assessing risks and planning responses in the European Union, May 2013. Euro surveillance: bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin. 2013:18
4. WHO. WHO RISK ASSESSMENT: Human infections with avian influenza A(H7N9) virus. 2014.
5. Miller RS, Sweeney SJ, Akkina JE, Saito EK. Potential Intercontinental Movement of Influenza A(H7N9) Virus into North America by Wild Birds: Application of a Rapid Assessment Framework. Transboundary and emerging diseases. 2014 doi:10.1111/tbed.12213
6. Olson SH, Gilbert M, Cheng MC, Mazet JA, Joly DO. Historical prevalence and distribution of avian influenza virus A(H7N9) among wild birds. Emerging infectious diseases. 2013;19:2031-3 doi:10.3201/eid1912.130649
7. The fight against bird flu. Nature. 2013;496:397
8. Andre FE, Booy R, Bock HL, Clemens J, Datta SK, John TJ. et al. Vaccination greatly reduces disease, disability, death and inequity worldwide. Bulletin of the World Health Organization. 2008;86:140-6
9. Rebmann T, Zelicoff A. Vaccination against influenza: role and limitations in pandemic intervention plans. Expert review of vaccines. 2012;11:1009-19 doi:10.1586/erv.12.63
10. Kumar S, Henrickson KJ. Update on influenza diagnostics: lessons from the novel H1N1 influenza A pandemic. Clinical microbiology reviews. 2012;25:344-61 doi:10.1128/CMR.05016-11
11. von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nature reviews Drug discovery. 2007;6:967-74 doi:10.1038/nrd2400
12. Du QS, Huang RB, Wang SQ, Chou KC. Designing inhibitors of M2 proton channel against H1N1 swine influenza virus. PloS one. 2010;5:e9388. doi:10.1371/journal.pone.0009388
13. TH M. Panel urges wide use of antiviral drug. Science. 1979;206:1058-60
14. Gubareva LV, Kaiser L, Hayden FG. Influenza virus neuraminidase inhibitors. Lancet. 2000;355:827-35 doi:10.1016/S0140-6736(99)11433-8
15. Furuse Y, Suzuki A, Kamigaki T, Oshitani H. Evolution of the M gene of the influenza A virus in different host species: large-scale sequence analysis. Virology journal. 2009;6:67. doi:10.1186/1743-422X-6-67
16. Furuse Y, Suzuki A, Oshitani H. Large-scale sequence analysis of M gene of influenza A viruses from different species: mechanisms for emergence and spread of amantadine resistance. Antimicrobial agents and chemotherapy. 2009;53:4457-63 doi:10.1128/AAC.00650-09
17. Abed Y, Goyette N, Boivin G. Generation and characterization of recombinant influenza A (H1N1) viruses harboring amantadine resistance mutations. Antimicrobial agents and chemotherapy. 2005;49:556-9 doi:10.1128/AAC.49.2.556-559.2005
18. Nelson MI, Simonsen L, Viboud C, Miller MA, Holmes EC. The origin and global emergence of adamantane resistant A/H3N2 influenza viruses. Virology. 2009;388:270-8 doi:10.1016/j.virol.2009.03.026
19. Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular basis of the specific anti-influenza action of amantadine. The EMBO journal. 1985;4:3021-4
20. Belshe RB, Smith MH, Hall CB, Betts R, Hay AJ. Genetic basis of resistance to rimantadine emerging during treatment of influenza virus infection. Journal of virology. 1988;62:1508-12
21. Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH. et al. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1315-20 doi:10.1073/pnas.1216526110
22. Hurt AC, Holien JK, Parker MW, Barr IG. Oseltamivir resistance and the H274Y neuraminidase mutation in seasonal, pandemic and highly pathogenic influenza viruses. Drugs. 2009;69:2523-31 doi:10.2165/11531450-000000000-00000
23. Baz M, Abed Y, Simon P, Hamelin ME, Boivin G. Effect of the neuraminidase mutation H274Y conferring resistance to oseltamivir on the replicative capacity and virulence of old and recent human influenza A(H1N1) viruses. The Journal of infectious diseases. 2010;201:740-5 doi:10.1086/650464
24. Hurt AC, Ernest J, Deng YM, Iannello P, Besselaar TG, Birch C. et al. Emergence and spread of oseltamivir-resistant A(H1N1) influenza viruses in Oceania, South East Asia and South Africa. Antiviral research. 2009;83:90-3 doi:10.1016/j.antiviral.2009.03.003
25. McKimm-Breschkin JL, Sahasrabudhe A, Blick TJ, McDonald M, Colman PM, Hart GJ. et al. Mutations in a conserved residue in the influenza virus neuraminidase active site decreases sensitivity to Neu5Ac2en-derived inhibitors. Journal of virology. 1998;72:2456-62
26. V. Karthick, Ramanathan K. Insight into the Oseltamivir Resistance R292K Mutation in H5N1 Influenza Virus: A Molecular Docking and Molecular Dynamics Approach. Cell Biochemistry and Biophysics. 2014;68:291-9
27. Kiso M, Mitamura K, Sakai-Tagawa Y, Shiraishi K, Kawakami C, Kimura K. et al. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet. 2004;364:759-65 doi:10.1016/S0140-6736(04)16934-1
28. Gillman A, Muradrasoli S, Soderstrom H, Nordh J, Brojer C, Lindberg RH. et al. Resistance mutation R292K is induced in influenza A(H6N2) virus by exposure of infected mallards to low levels of oseltamivir. PloS one. 2013;8:e71230. doi:10.1371/journal.pone.0071230
29. Wu Y, Bi Y, Vavricka CJ, Sun X, Zhang Y, Gao F. et al. Characterization of two distinct neuraminidases from avian-origin human-infecting H7N9 influenza viruses. Cell research. 2013;23:1347-55 doi:10.1038/cr.2013.144
30. Yen HL, McKimm-Breschkin JL, Choy KT, Wong DD, Cheung PP, Zhou J. et al. Resistance to neuraminidase inhibitors conferred by an R292K mutation in a human influenza virus H7N9 isolate can be masked by a mixed R/K viral population. mBio. 2013 4. doi:10.1128/mBio.00396-13
31. Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM. et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45-9 doi:10.1038/nature05114
32. Liu Q, Lu L, Sun Z, Chen GW, Wen Y, Jiang S. Genomic signature and protein sequence analysis of a novel influenza A (H7N9) virus that causes an outbreak in humans in China. Microbes and infection / Institut Pasteur. 2013;15:432-9 doi:10.1016/j.micinf.2013.04.004
33. Hai R, Schmolke M, Leyva-Grado VH, Thangavel RR, Margine I, Jaffe EL. et al. Influenza A(H7N9) virus gains neuraminidase inhibitor resistance without loss of in vivo virulence or transmissibility. Nature communications. 2013;4:2854. doi:10.1038/ncomms3854
34. Hay AJ, Hayden FG. Oseltamivir resistance during treatment of H7N9 infection. Lancet. 2013;381:2230-2 doi:10.1016/S0140-6736(13)61209-X
35. Itamura S. [Structure and function of influenza virus neuraminidase]. Nihon rinsho Japanese journal of clinical medicine. 1997;55:2570-4
36. Shtyrya YA, Mochalova LV, Bovin NV. Influenza virus neuraminidase: structure and function. Acta naturae. 2009;1:26-32
37. Xu X, Zhu X, Dwek RA, Stevens J, Wilson IA. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. Journal of virology. 2008;82:10493-501 doi:10.1128/JVI.00959-08
38. Babu YS, Chand P, Bantia S, Kotian P, Dehghani A, El-Kattan Y. et al. BCX-1812 (RWJ-270201): discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. Journal of medicinal chemistry. 2000;43:3482-6
39. Lew W, Chen X, Kim CU. Discovery and development of GS 4104 (oseltamivir): an orally active influenza neuraminidase inhibitor. Current medicinal chemistry. 2000;7:663-72
40. von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B. et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418-23 doi:10.1038/363418a0
41. Sugaya N, Ohashi Y. Long-acting neuraminidase inhibitor laninamivir octanoate (CS-8958) versus oseltamivir as treatment for children with influenza virus infection. Antimicrobial agents and chemotherapy. 2010;54:2575-82 doi:10.1128/AAC.01755-09
42. Berendsen HJC. et al. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995:43-56 doi:10.1016/0010-4655(95)00042-e
43. Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. Journal of Chemical Theory and Computation. 2008;4:435-47 doi:10.1021/ct700301q
44. Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. Journal of computational chemistry. 2004;25:1656-76 doi:10.1002/jcc.20090
45. Oostenbrink C, Soares TA, van der Vegt NF, van Gunsteren WF. Validation of the 53A6 GROMOS force field. European biophysics journal: EBJ. 2005;34:273-84 doi:10.1007/s00249-004-0448-6
46. Cino EA, Choy WY, Karttunen M. Comparison of Secondary Structure Formation Using 10 Different Force Fields in Microsecond Molecular Dynamics Simulations. J Chem Theory Comput. 2012;8:2725-40 doi:10.1021/ct300323g
47. Berendsen HJC, Postma JPM, Gunsteren WF, Hermans J. Interaction Models for Water in Relation to Protein Hydration. Intermolecular Forces. 1981;14:331-42 doi:10.1007/978-94-015-7658-1_21
48. Darden T, York D, Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics. 1993;98:10089. doi:10.1063/1.464397
49. Chen CY. TCM Database@Taiwan: the world's largest traditional Chinese medicine database for drug screening in silico. PloS one. 2011;6:e15939. doi:10.1371/journal.pone.0015939
50. Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug discovery today. 2006;11:580-94 doi:10.1016/j.drudis.2006.05.012
51. Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Current medicinal chemistry. 2013;20:2839-60
52. Sanner MF. A component-based software environment for visualizing large macromolecular assemblies. Structure. 2005;13:447-62 doi:10.1016/j.str.2005.01.010
53. Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity: a rapid access to atomic charges. Tetrahedron. 1980;36:3219-28
54. Sanner MF. Python: a programming language for software integration and development. Journal of molecular graphics & modelling. 1999;17:57-61
55. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry. 2009;30:2785-91 doi:10.1002/jcc.21256
56. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry. 2010;31:455-61 doi:10.1002/jcc.21334
57. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 2001;46:3-26
58. Kumari R, Kumar R, Open Source Drug Discovery C, Lynn A. g_mmpbsa-A GROMACS Tool for High-Throughput MM-PBSA Calculations. Journal of chemical information and modeling. 2014;54:1951-62 doi:10.1021/ci500020m
59. Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144-9
60. Nguyen HT, Fry AM, Gubareva LV. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antiviral therapy. 2012;17:159-73 doi:10.3851/IMP2067
61. Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W. et al. Human infection with a novel avian-origin influenza A (H7N9) virus. The New England journal of medicine. 2013;368:1888-97 doi:10.1056/NEJMoa1304459
Author contact
![]() Corresponding author: Email: ly.leedu.vn; Telephone: 084 (08)3715.4718; Fax: 084 (08) 3715.4719
Corresponding author: Email: ly.leedu.vn; Telephone: 084 (08)3715.4718; Fax: 084 (08) 3715.4719