Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(10):1073-1081. doi:10.7150/ijms.9282 This issue Cite
Research Paper
Astragaloside IV Alleviates Early Brain Injury Following Experimental Subarachnoid Hemorrhage in Rats
Anwen Shao1, Songxue Guo2, Sheng Tu3, Al-baadani Ammar1, Junjia Tang1, Yuan Hong1, Haijian Wu1, Jianmin Zhang1 ![]()
1. Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China;
2. Department of Burns, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China;
3. First College of Clinical Medicine, Zhejiang Chinese Medical University, Hangzhou, China.
Received 2014-4-2; Accepted 2014-7-1; Published 2014-8-8
Abstract
Astragaloside IV, one of the main effective components isolated from Astragalus membranaceus, has multiple neuroprotective properties, while the effects of astragaloside IV on the attenuation of subarachnoid hemorrhage (SAH)-induced early brain injury (EBI) and its possible mechanisms are unknown. In the present study, we aimed to determine whether astragaloside IV could inhibit oxidative stress, reduce neuronal apoptosis, and improve neurological deficits after experimental SAH in rats. Rats (n=68) were randomly divided into the following groups: Sham group, SAH group, SAH+vehicle group, and SAH+astragaloside IV group. Astragaloside IV or an equal volume of vehicle was administered at 1 h and 6 h after SAH, all the rats were subsequently sacrificed at 24 h after SAH. Mortality, neurological scores, and brain edema were assessed, biochemical tests and histological studies were also performed at that point. SAH induced an increase in the malondialdehyde (MDA) level, neuronal apoptosis, cleaved caspase 3, brain edema and decreased activities of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px). Astragaloside IV treatment reversed these changes and improved neurobehavioral outcomes of SAH rats. Our findings suggested that astragaloside IV may alleviate EBI after SAH through antioxidative and anti-apoptotic effects.
Keywords: astragaloside IV, oxidative stress, apoptosis, early brain injury, subarachnoid hemorrhage.
Introduction
While subarachnoid hemorrhage (SAH) accounts for only 5% of strokes [1], it is associated with high mortality and severe morbidity. Although medical technology and treatment modality continues to advance, the prognosis of SAH patients is still poor. Traditionally, delayed cerebral vasospasm, which defined as a pathological demonstration occurred in the late phase of SAH (3-7 days), was considered as the most important cause of delayed ischemic neurological deficits after SAH onset. To date, tremendous research efforts have been made to reduce SAH-induced cerebral vasospasm [2-4]. Although positive results were obtained from animal experiments [5-7], however, anti-vasospasm treatments were failed to improve the SAH patients' outcome in most clinical trials [8]. These results from clinical trials make researchers raise doubt on the role of vasospasm, and attempt finding new targets in treating patients suffering from SAH. Early brain injury (EBI), which firstly proposed by Kusaka et al. in 2004, refers to a pathophysiological process occurring within the first 72 h after SAH. Accumulating evidences indicate that EBI plays a significant role in patients' neurological deficits and poor outcome after SAH [9]. Noteworthily, pathological factors, such as oxidative stress and apoptosis, are involved in the pathogenesis of SAH-induced EBI [1, 10, 11]. Generation of oxygen free radicals leads to lipid peroxidation, which could result in brain damage after SAH. On the other hand, the antioxidant defense system, which consists of superoxide dismutase (SOD), glutathione peroxidase (GSH-PX) and other antioxidant enzymes, is inhibited and inefficient to scavenge excessive oxygen free radicals following SAH, which resultantly exacerbates the severity of SAH-induced brain injury [12]. Cell apoptosis, another significant pathological event, has also acquired much attention due to its importance in EBI [13]. SAH could induce apoptosis in different cell types, icluding neurons, astrocytes and oligodendrocytes [14], among them, apoptotic cell death of neurons has a close relationship with neurobehavior functions after SAH [10, 14, 15]. Therefore, therapies targeting these early pathophysiological processes may inhibit EBI and reduce the incidence of subsequent neurological complications following SAH.
Astragaloside IV (Fig. 1) is the main effective components isolated from Astragalus membranaceus [16, 17], and widely used in treating various diseases in traditionally Chinese medicine [18-21]. Of note, the neuroprotective effects of astragaloside IV have been demonstrated previously in a variety of central nervous system injuries, including cerebral ischemic-reperfusion injury, Parkinson's disease, et al [22-25]. The favorable effects of astragaloside IV are associated with its multiple properties including anti-oxidant [26], anti-apoptosis [27], anti-inflammation [28], immune-enhancement [29]. However, the effects of astragaloside IV on SAH-induced EBI have not been investigated. Moreover, the molecular mechanisms underlying astragaloside IV-mediated neuroprotection is less elucidated.
Chemical structure of astragaloside IV.
In the current work, we hypothesized that astragaloside IV might attenuate EBI and improve neurological outcomes by alleviating oxidative stress and neuronal apoptosis following SAH.
Materials and methods
Animals
Male Sprague-Dawley rats (280-350 g) were obtained from the Animal Center of Zhejiang Chinese Medical University (Hangzhou, China) and were randomly divided into four groups: rats in the sham group (n=17) underwent a procedure similar to that of the SAH group except perforation; rats in the SAH group (n=17) underwent SAH without any treatment; rats in SAH+vehicle group (n=17) underwent SAH and were treated with physiological saline; and rats in the SAH+astragaloside IV group (n=17) underwent SAH and were treated with 10 mg/kg astragaloside IV. All parameters were investigated at 24 h after SAH (Fig. 2). All experimental protocols were approved by the Ethics Committee for the Use of Experimental Animals of Zhejiang University.
Experimental design and animal group classification.
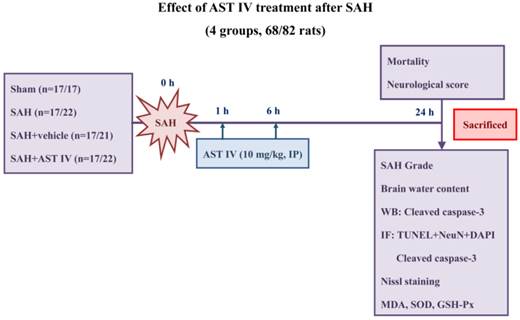
Rat SAH model and mortality
SAH was performed by using an endovascular perforation method as described previously, with small modification [30]. Briefly, animals were anesthetized with an intraperitoneal injection of chloral hydrate (400 mg/kg). The rectal temperature was maintained at 37°C with a heating blanket during the operation. The left common, external and internal carotid arteries were exposed, a sharpened 4-0 monofilament nylon suture was inserted into the left internal carotid artery through the external carotid artery to perforate the artery at the bifurcation of the anterior and middle cerebral artery and cause SAH. Sham-operated rats underwent similar procedures except the perforation. All animals were kept in an air-conditioned room with temperature maintained at 27°C, and the rats were allowed access to food and water ad libitum before and after surgery. Mortality was calculated 24 h after SAH.
Drug administration
The purity of astragaloside IV (Sobeo Biotech Co., Xi'an, ShangXi, China) was evaluated via HPLC analysis, and high purity (99%) astragaloside IV was dissolved in physiological saline to a storage concentration of 1.0 mg/ml, containing 5% alcohol (5:95, v/v), as previously described [24]. The injection of astragaloside IV (10 mg/kg, 3 ml) was given intraperitoneally at 1 h and 6 h after SAH. The dose selection is based on previous studies [24]. The SAH+vehicle group rats were injected with an identical volume of physiological saline containing 5% alcohol.
Assessment of SAH grade and neurological score
As a parameter to evaluate the severity of SAH, SAH grade was obtained according to a grading system that was described previously [31]. Briefly, the system was based on the amount of subarachnoid blood clots distributed in the six segments of basal cistern: grade 1, no subarachnoid blood (scores=0); grade 2, minimal subarachnoid clots (scores=1); grade 3, moderate subarachnoid clots with recognizable arteries (scores=2); and grade 4, blood clots covering all arteries (scores=3). A total score ranging from 0 to 18 was obtained by adding the scores from all 6 segments. The grading of SAH was performed by a partner who was blinded to the experiment.
Neurobehavioral scores were recorded at 24 h after SAH according to the Garcia's method [32, 33] by a partner who was blinded to the experiment. Briefly, an 18 point scoring system was used to assess animals' neurological deficits by six aspects, including spontaneous activity (0-3), symmetry in the movement of all limbs (0-3), forepaw outstretching (0-3), climbing (1-3), body proprioception (1-3) and response to vibrissae touch (1-3). The minimum neurological score was 3 and the maximum was 18; a lower score represents serious neurological deficits.
Evaluation of brain water content
Rats (n=5) were sacrificed at 24 h after SAH. The brain was rapidly removed and weighed immediately (wet weight). The brain samples were dried in an oven at 105 ◦C for 24 h and weighed again (dry weight). The brain water content was calculated as [(wet weight- dry weight)/wet weight] ×100% [34].
Measurement of lipid peroxidation and antioxidant enzymatic activities
Rats (n=6) were sacrificed at 24 h after SAH induction, left (injured side) basal cortical samples were obtained for assessment as previously described [6]. Lipid peroxidation was assessed by measurement of malondialdehyde (MDA) at 24 h after SAH. MDA content was measured by commercial kits (Beyotime, Haimen, Jiangsu, China), according to the methods described by the assay kits. The absorbance of the supernatant was measured by spectrophotometry at 532 nm. All tests were conducted in triplicate. The MDA concentration was calculated from the standard curve and expressed as nmol MDA/mg protein. SOD and GSH-Px which are endogenous scavengers of reactive oxygen species (ROS), were measured in brain tissue using commercial assay kits (Cayman Chemical, Ann Arbor, MI, USA), according to the manufacturer's instructions. Assessment of SOD activity was based on its properties of inhibition of superoxide radicals produced by xanthine oxidase system, and SOD levels were expressed as units/mg protein. The GSH-Px activity was determined following the manufacturer's instructions. Briefly, GSH-Px activity was indirectly measured by a coupled reaction with glutathione reductase (GR). Oxidized glutathione (GSSG), which is produced upon reduction of ROOH by GSH-Px was restored to its reduced state by GR and Nicotinamide Adenine Dinucleotide Phosphate (NADPH). GR decreased NADPH to NADP+, leading to a reduction in absorbance at 340 nm. One unit of GSH-Px was defined as the amount of enzyme that catalyzed the oxidation of 1.0mmol of NADPH to NADP+ per minute at 25°C. GSH-Px activities of the brain samples were expressed as U/mg protein.
Double-Label Staining of TUNEL and NeuN
Rats (n=6) were decapitated at 24 h following SAH injury, and left (injured side) basal cortical samples were taken for double label staining. Double staining of Terminal-deoxynucleoitidyl Transferase Mediated Nick End Labeling (TUNEL) and neuronal marker (NeuN) was performed to confirm the colocalization of apoptotic cells and neurons. At 24 h after SAH, rats were sacrificed and perfused with 4% paraformaldehyde. Brains were postfixed in the same fixative for 1 d and then paraffin-embedded. Then, the brains were sectioned at a 10μm thickness. The sections were reacted with the antibody against NeuN (Abcam, Cambridge, MA, USA) and, then, subjected to TUNEL staining (In Situ Cell Death Detection kit, Roche, Mannheim, Germany). Finally, the sections were covered with 4'6-diamidino-2-phenylindole (DAPI) (Beyotime) and observed under a fluorescent microscope (Leica, Germany).
Assay of Nissl staining
SAH-induced neuron death was assessed by Nissl staining. Neurons with round and palely stained nuclei were regarded as surviving cells, while shrunken neurons with condensed nuclei were regarded as damaged cells. Nissl staining was performed according to the manufacturer's protocol (Roche), and tissues were examined under a light microscope by a partner blind to the grouping.
Immunofluorescence staining of caspase 3
At 24 h after SAH, rats were decapitated and perfused with PBS and 4% paraformaldehyde. Brains were placed in the same fixative for 1 d and paraffin-embedded. Then, the brains were cut in 10μm thickness. The immunofluorescence staining was performed as described previously [35], and observed under a fluorescent microscope.
Western blot
Rats (n=6) were decapitated at 24 h after SAH induction or sham operation. The same part of the left basal cortical sample was obtained, and whole cell protein extraction was performed as previously described [35]. Briefly, the samples were homogenized and centrifuged at 12,000 × g for 10 min at 4 °C. Supernatants were collected, and protein concentrations were determined with a BCA kit (Beyotime). The protein samples were separated by 10-15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, Temecula, CA, USA). After blocking with 5% nonfat dry milk in TBS for 2 h, we incubated the membranes overnight at 4 °C with primary antibodies for cleaved caspase-3 (dilution 1:500, Abcam), β-actin (dilution 1:1000, Abcam). After incubation, the membranes were washed with TBST and incubated with horseradish-peroxidase conjugated secondary antibodies for 2 h at room temperature. We detected the antigen-antibody complexes using an ECL Plus chemiluminescence reagent kit (Beyotime) and exposed them to X-ray film. Band densities were quantified by Image J software (NIH).
Statistical analysis
Values were expressed as the means ± SD. A one-way analysis of variance (ANOVA) was used for comparisons between multiple groups, followed by Tukey's test. For mortality, Fisher's exact test was used for group comparisons. P< 0.05 was considered statistically significant.
Results
Mortality, neurological score and SAH grade at 24 h after SAH
None of rats in the Sham group died (0 of 17 rats). Mortality was 22.7% (5 of 22 rats) in the SAH group, 19.0% (4 of 21 rats) in the SAH+vehicle group and 22.7% (5 of 22 rats) in the SAH+astragaloside IV group (Fig. 3B). Most of them died within 3 hours after SAH. The dead rats were excluded from further analysis. The SAH grade score had no significant difference between SAH operated groups; Astragaloside IV did not alter the bleeding level compared to other two groups (n=17, P> 0.05, Fig. 3C). The neurological score was significantly decreased in the SAH group compared to the Sham group (n=17, P< 0.05, Fig. 3D). After astragaloside IV treatment, neurological deficits were reduced compared to vehicle treatment (n=17, P< 0.05, Fig. 3D).
(A) Representative photos of rat brains after surgery. The same part of basal cortical brain tissue was taken for tests (circled areas). (B) Mortality was not significantly different among SAH groups (P> 0.05). (C) Summary of SAH grade in different groups (n=17). (D) Neurological score decreased markedly in SAH group (n=17, P< 0.05) and increased after administration of astragaloside IV (n=17, P< 0.05). (E) Brain water content increased significantly after SAH and decreased with astragaloside IV treatment (n=5, P< 0.05). *P< 0.05 compared with sham group; **P< 0.05 compared with SAH+vehicle group.
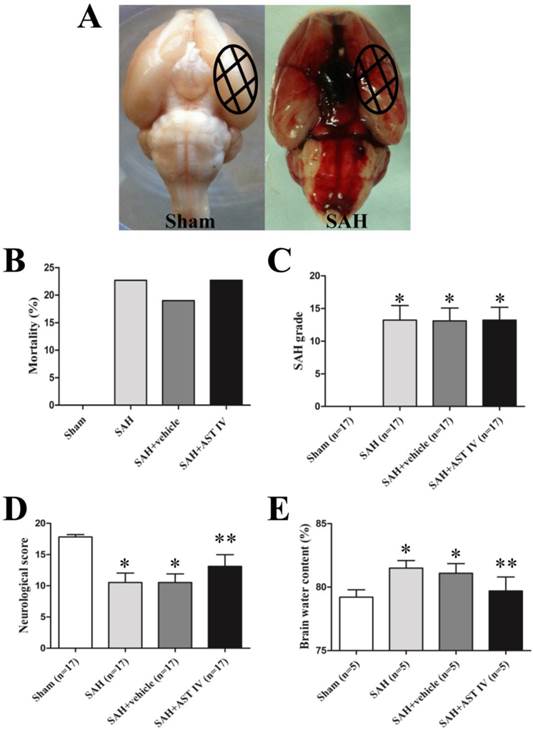
Astragaloside IV attenuated brain edema at 24 h after SAH
As show in Fig. 3E, brain water content was markedly higher in SAH and SAH+vehicle groups compared to the sham group at 24 h after SAH (n=5, P< 0.05). Brain edema was attenuated significantly after administration of astragaloside IV (n=5, P< 0.05).
Astragaloside IV reduced lipid peroxidation and improved antioxidant enzymatic activity at 24 h after SAH
Obvious up-regulation of MDA and down-regulation of the activities of SOD and GSH-Px were observed in the SAH and SAH+vehicle groups compared with Sham group (n=6, P< 0.05, Fig. 4A-C). Injection of astragaloside IV significantly reduced the level of MDA and elevated the activities of antioxidant enzymes at 24 h after SAH (n=6, P< 0.05, Fig. 4A-C).
MDA (A) elevated markedly in SAH and SAH+vehicle groups compared to sham group, and was reversed by astragaloside IV treatment (n=6, P< 0.05). GSH-Px (B) and SOD (C) activities reduced obviously after SAH and increased after astragaloside IV treatment (n=6, P< 0.05). (D) Immunofluorence of cleaved caspase 3. (E,F). Western blot analysis of cleaved caspase 3 in the basal cortex of left hemisphere (n=6, P< 0.05). *P< 0.05 compared with sham group; **P< 0.05 compared with SAH+vehicle group.
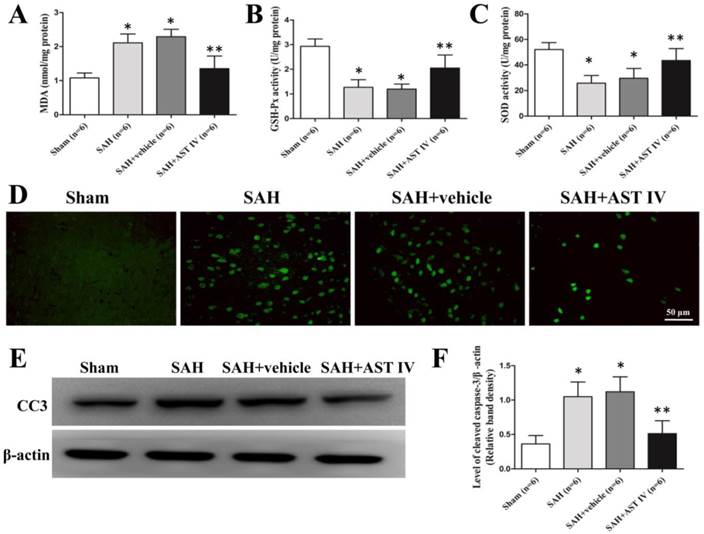
Astragaloside IV down-regulated the protein expression of cleaved caspase 3 at 24 h after SAH
The same part of the left basal cortex was taken for detection of cleaved caspase 3. The protein level of cleaved caspase 3 was low in the sham group, decreased markedly in the SAH and SAH+vehicle groups and increased markedly in the SAH+astragaloside IV group (n=6, P< 0.05, Fig. 4E,F). Cleaved caspase 3 examined by immunofluorescence showed a similar trend (n=6, Fig. 4D).
Astragaloside IV inhibited neuronal death in the cortex at 24 h after SAH
Fig. 5A shows almost no TUNEL-positive cells were observed in Sham group. On the contrary, numerous TUNEL-positive cells were detected at 24 h after SAH (n=6, P< 0.05), and the astragaloside IV-treated group had a substantial increase in the number of surviving cells compared to SAH and SAH+vehicle groups (n=6, P< 0.05). Most TUNEL-positive cells were colocalized with neurons, which indicates that cell death occurs mainly in neurons at 24 h after SAH.
(A) Double staining of TUNEL (green) and NeuN (red); nuclei were counterstained with DAPI (blue). TUNEL-positive cells mainly colocalized with neurons. Massive numbers of apoptotic cells were observed in SAH and SAH+vehivle groups; Astragaloside IV inhibited apoptosis significantly compared to SAH groups (n=6). Nissl staining (B) and quantitative analysis (C) showed that neurons were normal with sharp demarcations in the sham group. Damaged neurons were present with deformations and condensation of cytoplasm and nuclei in SAH and SAH+vehicle groups, and this was attenuated by treatment with astragaloside IV (n=6).
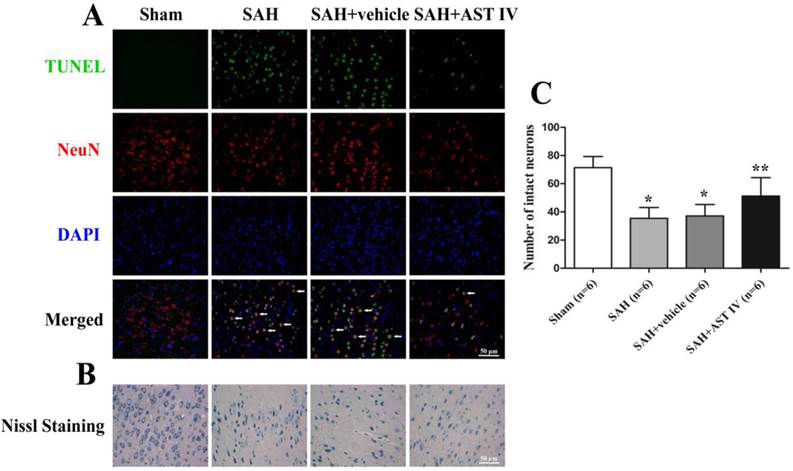
Astragaloside IV attenuated neuronal injury in the cortex at 24 h after SAH
As demonstrated in Fig. 5B,C, no evident injury of neurons was found in the Sham group, while in the SAH and SAH+vehicle groups, neurons were obviously damaged (deformation and condensation of cytoplasm and nucleus). Neuron injury was significantly alleviated by treatment with astragaloside IV (n=6).
Discussion
In the past decades, delayed cerebral vasospasm was thought to be the key factor leading to high mortality and morbidity after SAH [36]. Nevertheless, the failure of anti-vasospastic drugs to improve outcome in clinical trials has led researchers to cast doubt on the significance of vasospasm and to seek for new concepts in the treatment of SAH [11, 37-42]. Currently, accumulating evidences demonstrated that EBI tightly correlates with the prognosis of SAH patients [10]. Various pathological mechanisms, including oxidative stress and apoptotic cell death, participate in the pathogenesis of EBI [11]. And therapeutic approaches targeting for SAH-induced EBI is protective [6, 34, 43, 44]. In this study, we explored the damage mechanisms of EBI, and evaluate the potential beneficial effects of astragaloside IV on EBI. We found that astragaloside IV could reduce oxidative stress, inhibit apoptotic cell death, attenuate brain edema and finally alleviate neurological deficits following SAH.
Oxidative stress, which closely associated with the over-production of free radical, including reactive oxygen species, participates in various neurological diseases [22, 45, 46]. A number of evidence has demonstrated that the imbalance of free radical and the antioxidative defense system, has led to the occurrence of oxidative stress. Previous studies have shown that astragaloside IV acts as an efficient free radical scavenger and strengthens the intrinsic anti-oxidization system in the animal model of cerebral ischemia/reperfusion [22]. In this study, we found that astragaloside IV alleviated SAH-induced oxidative stress injury by eliminating ROS and strengthening the activities of antioxidases.
Apoptotic cell death of neurons after SAH has an important effect on the neurobehavioral outcome after SAH [47]. Notably, the caspase-dependent pathway plays a central role in mediating cell apoptosis [11, 47]. Inhibition of caspase-dependent apoptosis of neurons contributes to attenuate SAH-induced EBI [15, 43]. On the other hand, Dingkun Gu et al. reported that astragaloside IV could reduce glucose-induced apoptosis in podocyte via down-regulation of caspase 3 [48]. Besides, Zhang Q et al. suggested that astragaloside IV could reduce hypoxia-induced hippocampal neuronal apoptosis by decreasing the expression of hypoxia-inducible factor-1α (HIF-1α) [49]. Consistent with previous studies, we found that treatment with astragaloside IV suppressed caspase-dependent apoptosis in neurons, even in the setting of SAH.
Brain edema is an independent risk factor for mortality and poor outcome after SAH. Attenuation of brain edema is helpful to improve poor neurological outcome of neurological diseases, including SAH [50-52]. Of interest, Y.Z. Qu et al. found that astragaloside IV has the ability to attenuate brain edema by maintaining the integrity of BBB after cerebral ischemia/reperfusion injury [24]. In current study, we also found that astragaloside IV treatment has beneficial effects in reducing brain edema after SAH.
Interestingly, although we observed that astragaloside IV exert anti-oxidative stress and anti-apoptosis effect in our experiment, we found no significant difference in mortality at 24 h after SAH induction between SAH group and SAH+astragaloside IV group, which also seen in some previous studies using other drugs [32, 53-56]. This may due to the limitation of dose response and time window of the astragaloside IV. Therefore, the optimal time window and dosage, the appropriate injection route needs to be addressed in our further study. Additionally, some unknown potential mechanism which contributes to the adverse effect of astragaloside IV, may counteract the beneficial effect of astragaloside IV, and ultimately leads to make the mortality no statistical difference. Hence, whether astragaloside IV have side effects which may neutralize its neuroprotective effect and the underlying mechanism should be further studied.
In the present study, astragaloside IV appears to have a multiple beneficial properties in subarachnoid hemorrhage. However, the potential mechanism underlying astragaloside IV-induced neuroprotective effect still remains obscure. According to the results of previous literatures and our investigations, we speculated that the positive effect may due to the factors as following: Firstly, oxidative stress induced by SAH was the key factor that leads to brain injury[57], and numerous studies have shown that astragaloside IV could inhibit oxidative damage by scavenge free radicals and enhance antioxidant system [21, 25, 26, 58], and this process may associate with Nrf2 signaling pathway[59]. In terms of our understanding, the anti-oxidative stress ability of astragaloside IV may play a vital role in alleviating SAH-induced injury. Furthermore, both animal and clinical studies have shown that the excessive production of ROS promotes apoptosis in SAH, and multiple apoptotic signaling pathway were activated, including caspase-dependent pathway [5], caspase-independent pathway [60], mitochondrial pathway [61] and the death receptor pathway [62]. Therefore, we conjecture that the anti-apoptosis effect of astragaloside IV may partially be a result of oxidative stress reduction. There are many previous articles have demonstrated that astragaloside IV could inhibit apoptosis in multiple pathway, such as Fas/Fas L apoptotic pathway [27], MAPK pathway [63], PI3K/Akt pathway [64], Bax-mediated pathway [25]. In our experiment, we have not explore the exact apoptosis signaling pathway involving in the astragaloside IV's anti-apoptotic effect, based on the results of caspase 3, we can only conclude that caspase-dependent pathway may involve, and the more precise upstream pathway needs to be elucidated in our future study. What's more, the effect of astragaloside IV in reducing brain edema may associated with apoptosis inhibition and oxidative stress reduction after SAH, as previous studies have revealed that both oxidative injury and apoptosis are taking part in blood-brain barrier disruption, resultantly lead to brain edema [15, 65]. Taken together, we speculate that inhibits lipid peroxidation, stimulates GSH-Px activity, improves SOD activity are the primary effects of astragaloside IV, but suppresses neuronal apoptosis and reduces brain edema may be the secondary effects of astragaloside IV, which may partially be the consequence of oxidative injury improvement.
In summary, the current work demonstrated that astragaloside IV significantly inhibited oxidative stress, suppressed neuronal apoptosis, reduced brain edema, and attenuated neurological deficits at 24 h after SAH. Noteworthily, this is the first study to investigate the effects of astragaloside IV treatment after SAH. The best time window, the suitable route of administration, and exact mechanisms of astragaloside IV in SAH treatment, however, are warranted to be further elucidated.
Acknowledgements
This work was supported by Grant 81171096, 81371369, 81371433 from National Natural Science Foundation of China; Grant 20120101120030 from Doctoral Program of the Ministry of Education; Grant 2013KYA088 from Health department of Zhejiang province.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen S, Feng H, Sherchan P. et al. Controversies and evolving new mechanisms in subarachnoid hemorrhage. Progress in neurobiology. 2014;115:64-91
2. Velat GJ, Kimball MM, Mocco JD, Hoh BL. Vasospasm after aneurysmal subarachnoid hemorrhage: review of randomized controlled trials and meta-analyses in the literature. World neurosurgery. 2011;76:446-54
3. Golan E, Vasquez DN, Ferguson ND. et al. Prophylactic magnesium for improving neurologic outcome after aneurysmal subarachnoid hemorrhage: systematic review and meta-analysis. Journal of critical care. 2013;28:173-81
4. Ma J, Huang S, Ma L. et al. Endothelin-receptor antagonists for aneurysmal subarachnoid hemorrhage: an updated meta-analysis of randomized controlled trials. Critical care. 2012;16:R198
5. Cheng G, Wei L, Zhi-Dan S. et al. Atorvastatin ameliorates cerebral vasospasm and early brain injury after subarachnoid hemorrhage and inhibits caspase-dependent apoptosis pathway. BMC neuroscience. 2009;10:7
6. Hong Y, Guo S, Chen S. et al. Beneficial effect of hydrogen-rich saline on cerebral vasospasm after experimental subarachnoid hemorrhage in rats. Journal of neuroscience research. 2012;90:1670-80
7. Sugawara T, Ayer R, Jadhav V. et al. Simvastatin attenuation of cerebral vasospasm after subarachnoid hemorrhage in rats via increased phosphorylation of Akt and endothelial nitric oxide synthase. Journal of neuroscience research. 2008;86:3635-43
8. Wang X, Li YM, Li WQ. et al. Effect of clazosentan in patients with aneurysmal subarachnoid hemorrhage: a meta-analysis of randomized controlled trials. PloS one. 2012;7:e47778
9. Kusaka G, Ishikawa M, Nanda A. et al. Signaling pathways for early brain injury after subarachnoid hemorrhage. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2004;24:916-25
10. Caner B, Hou J, Altay O. et al. Transition of research focus from vasospasm to early brain injury after subarachnoid hemorrhage. Journal of neurochemistry. 2012;123(Suppl 2):12-21
11. Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Progress in neurobiology. 2012;97:14-37
12. Ayer RE, Zhang JH. Oxidative stress in subarachnoid haemorrhage: significance in acute brain injury and vasospasm. Acta neurochirurgica Supplement. 2008;104:33-41
13. Yuksel S, Tosun YB, Cahill J, Solaroglu I. Early brain injury following aneurysmal subarachnoid hemorrhage: emphasis on cellular apoptosis. Turkish neurosurgery. 2012;22:529-33
14. Prunell GF, Svendgaard NA, Alkass K, Mathiesen T. Delayed cell death related to acute cerebral blood flow changes following subarachnoid hemorrhage in the rat brain. Journal of neurosurgery. 2005;102:1046-54
15. Park S, Yamaguchi M, Zhou C. et al. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35:2412-7
16. Li M, Wang W, Xue J. et al. Meta-analysis of the clinical value of Astragalus membranaceus in diabetic nephropathy. Journal of ethnopharmacology. 2011;133:412-9
17. Matkovic Z, Zivkovic V, Korica M. et al. Efficacy and safety of Astragalus membranaceus in the treatment of patients with seasonal allergic rhinitis. Phytotherapy research. 2010;24:175-81
18. Tu L, Pan CS, Wei XH. et al. Astragaloside IV protects heart from ischemia and reperfusion injury via energy regulation mechanisms. Microcirculation. 2013;20:736-47
19. Tan S, Wang G, Guo Y. et al. Preventive Effects of a Natural Anti-Inflammatory Agent, Astragaloside IV, on Ischemic Acute Kidney Injury in Rats. Evidence-based complementary and alternative medicine. 2013;2013:284025
20. Li M, Ma RN, Li LH. et al. Astragaloside IV reduces cerebral edema post-ischemia/reperfusion correlating the suppression of MMP-9 and AQP4. European journal of pharmacology. 2013;715:189-95
21. Gui D, Huang J, Liu W. et al. Astragaloside IV prevents acute kidney injury in two rodent models by inhibiting oxidative stress and apoptosis pathways. Apoptosis. 2013;18:409-22
22. Luo Y, Qin Z, Hong Z. et al. Astragaloside IV protects against ischemic brain injury in a murine model of transient focal ischemia. Neuroscience letters. 2004;363:218-23
23. Yang J, Li J, Lu J. et al. Synergistic protective effect of astragaloside IV-tetramethylpyrazine against cerebral ischemic-reperfusion injury induced by transient focal ischemia. Journal of ethnopharmacology. 2012;140:64-72
24. Qu YZ, Li M, Zhao YL. et al. Astragaloside IV attenuates cerebral ischemia-reperfusion-induced increase in permeability of the blood-brain barrier in rats. European journal of pharmacology. 2009;606:137-41
25. Zhang ZG, Wu L, Wang JL. et al. Astragaloside IV prevents MPP(+)-induced SH-SY5Y cell death via the inhibition of Bax-mediated pathways and ROS production. Molecular and cellular biochemistry. 2012;364:209-16
26. He Y, Du M, Gao Y. et al. Astragaloside IV attenuates experimental autoimmune encephalomyelitis of mice by counteracting oxidative stress at multiple levels. PloS one. 2013;8:e76495
27. Kim MH, Kim SH, Yang WM. Beneficial effects of Astragaloside IV for hair loss via inhibition of Fas/Fas L-mediated apoptotic signaling. PloS one. 2014;9:e92984
28. Wang B, Chen MZ. Astragaloside IV possesses antiarthritic effect by preventing interleukin 1beta-induced joint inflammation and cartilage damage. Archives of pharmacal research. 2014;37:793-802
29. Hong F, Xiao W, Ragupathi G. et al. The known immunologically active components of Astragalus account for only a small proportion of the immunological adjuvant activity when combined with conjugate vaccines. Planta medica. 2011;77:817-24
30. Bederson JB, Germano IM, Guarino L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke. 1995;26:1086-91 discussion 91-2
31. Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. Journal of neuroscience methods. 2008;167:327-34
32. Chen S, Ma Q, Krafft PR. et al. P2X7 receptor antagonism inhibits p38 mitogen-activated protein kinase activation and ameliorates neuronal apoptosis after subarachnoid hemorrhage in rats. Critical care medicine. 2013;41:e466-74
33. Chen S, Ma Q, Krafft PR. et al. P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiology of disease. 2013;58:296-307
34. Altay O, Suzuki H, Hasegawa Y. et al. Isoflurane attenuates blood-brain barrier disruption in ipsilateral hemisphere after subarachnoid hemorrhage in mice. Stroke. 2012;43:2513-6
35. Hong Y, Shao A, Wang J. et al. Neuroprotective Effect of Hydrogen-Rich Saline against Neurologic Damage and Apoptosis in Early Brain Injury following Subarachnoid Hemorrhage: Possible Role of the Akt/GSK3beta Signaling Pathway. PloS one. 2014;9:e96212
36. Zoerle T, Ilodigwe D, Wan H. et al. Pharmacologic reduction of angiographic vasospasm in experimental subarachnoid hemorrhage: systematic review. Acta neurochirurgica Supplement. 2013;115:247-51
37. Kramer A, Fletcher J. Do endothelin-receptor antagonists prevent delayed neurological deficits and poor outcomes after aneurysmal subarachnoid hemorrhage? a meta-analysis. Stroke. 2009;40:3403-6
38. Macdonald RL, Higashida RT, Keller E. et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet neurology. 2011;10:618-25
39. Macdonald RL, Kassell NF, Mayer S. et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39:3015-21
40. Shaw MD, Vermeulen M, Murray GD. et al. Efficacy and safety of the endothelin, receptor antagonist TAK-044 in treating subarachnoid hemorrhage: a report by the Steering Committee on behalf of the UK/Netherlands/Eire TAK-044 Subarachnoid Haemorrhage Study Group. Journal of neurosurgery. 2000;93:992-7
41. Vajkoczy P, Meyer B, Weidauer S. et al. Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. Journal of neurosurgery. 2005;103:9-17
42. Macdonald RL, Higashida RT, Keller E. et al. Randomised trial of clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid hemorrhage undergoing surgical clipping (CONSCIOUS-2). Acta neurochirurgica Supplement. 2013;115:27-31
43. Wang Z, Ma C, Meng CJ. et al. Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. Journal of pineal research. 2012;53:129-37
44. Sherchan P, Lekic T, Suzuki H. et al. Minocycline improves functional outcomes, memory deficits, and histopathology after endovascular perforation-induced subarachnoid hemorrhage in rats. Journal of neurotrauma. 2011;28:2503-12
45. Cornelius C, Crupi R, Calabrese V. et al. Traumatic brain injury: oxidative stress and neuroprotection. Antioxidants & redox signaling. 2013;19:836-53
46. Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxidative medicine and cellular longevity. 2012;2012:428010
47. Hasegawa Y, Suzuki H, Sozen T. et al. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta neurochirurgica Supplement. 2011;110:43-8
48. Gui D, Guo Y, Wang F. et al. Astragaloside IV, a novel antioxidant, prevents glucose-induced podocyte apoptosis in vitro and in vivo. PloS one. 2012;7:e39824
49. Zhang Q, Gao WY, Zhang Y. et al. Protective effects of astragalus extract against intermittent hypoxia-induced hippocampal neurons impairment in rats. Chinese medical journal. 2013;126:1551-4
50. Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323-8
51. Xu M, Su W, Xu QP. Aquaporin-4 and traumatic brain edema. Chinese journal of traumatology. 2010;13:103-10
52. Shao AW, Wu HJ, Chen S. et al. Resveratrol attenuates early brain injury after subarachnoid hemorrhage through inhibition of NF-kappaB-dependent inflammatory/MMP-9 pathway. CNS neuroscience & therapeutics. 2014;20:182-5
53. Wu C, Hu Q, Chen J. et al. Inhibiting HIF-1alpha by 2ME2 ameliorates early brain injury after experimental subarachnoid hemorrhage in rats. Biochemical and biophysical research communications. 2013;437:469-74
54. Zhan Y, Chen C, Suzuki H. et al. Hydrogen gas ameliorates oxidative stress in early brain injury after subarachnoid hemorrhage in rats. Critical care medicine. 2012;40:1291-6
55. Altay O, Hasegawa Y, Sherchan P. et al. Isoflurane delays the development of early brain injury after subarachnoid hemorrhage through sphingosine-related pathway activation in mice. Critical care medicine. 2012;40:1908-13
56. Chen D, Wei XT, Guan JH. et al. Inhibition of c-Jun N-terminal kinase prevents blood-brain barrier disruption and normalizes the expression of tight junction proteins clautin-5 and ZO-1 in a rat model of subarachnoid hemorrhage. Acta neurochirurgica. 2012;154:1469-76 discussion 76
57. Gaetani P, Pasqualin A, Rodriguez y Baena R. et al. Oxidative stress in the human brain after subarachnoid hemorrhage. Journal of neurosurgery. 1998;89:748-54
58. Qi W, Niu J, Qin Q. et al. Astragaloside IV attenuates glycated albumin-induced epithelial-to-mesenchymal transition by inhibiting oxidative stress in renal proximal tubular cells. Cell stress & chaperones. 2014;19:105-14
59. Li X, Wang X, Han C. et al. Astragaloside IV suppresses collagen production of activated hepatic stellate cells via oxidative stress-mediated p38 MAPK pathway. Free radical biology & medicine. 2013;60:168-76
60. Endo H, Nito C, Kamada H. et al. Reduction in oxidative stress by superoxide dismutase overexpression attenuates acute brain injury after subarachnoid hemorrhage via activation of Akt/glycogen synthase kinase-3beta survival signaling. Journal of cerebral blood flow and metabolism. 2007;27:975-82
61. Jing CH, Wang L, Liu PP. et al. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012;213:144-53
62. Zhou C, Yamaguchi M, Kusaka G. et al. Caspase inhibitors prevent endothelial apoptosis and cerebral vasospasm in dog model of experimental subarachnoid hemorrhage. Journal of cerebral blood flow and metabolism. 2004;24:419-31
63. Wang Q, Shao X, Xu W. et al. Astragalosides IV inhibits high glucose-induced cell apoptosis through HGF activation in cultured human tubular epithelial cells. Renal failure. 2014;36:400-6
64. Jia Y, Zuo D, Li Z. et al. Astragaloside IV inhibits doxorubicin-induced cardiomyocyte apoptosis mediated by mitochondrial apoptotic pathway via activating the PI3K/Akt pathway. Chemical & pharmaceutical bulletin. 2014;62:45-53
65. Yan J, Li L, Khatibi NH. et al. Blood-brain barrier disruption following subarchnoid hemorrhage may be faciliated through PUMA induction of endothelial cell apoptosis from the endoplasmic reticulum. Experimental neurology. 2011;230:240-7
Author contact
![]() Corresponding author: Dr. Jianmin Zhang, Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China. Tel: +8613805722695. E-mail: zjm135sina.com, 21118116edu.cn.
Corresponding author: Dr. Jianmin Zhang, Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China. Tel: +8613805722695. E-mail: zjm135sina.com, 21118116edu.cn.