Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(10):1029-1038. doi:10.7150/ijms.8895 This issue Cite
Research Paper
Three-Dimensional Culture Environment Increases the Efficacy of Platelet Rich Plasma Releasate in Prompting Skin Fibroblast Differentiation and Extracellular Matrix Formation
Hussin A. Rothan1 ![]() , Ivan Djordjevic2, Hirbod Bahrani1, Mohammadjavad Paydar3, Fatimah Ibrahim2, Noorsaadah Abd Rahmanh4, Rohana Yusof1
, Ivan Djordjevic2, Hirbod Bahrani1, Mohammadjavad Paydar3, Fatimah Ibrahim2, Noorsaadah Abd Rahmanh4, Rohana Yusof1
1. Department of Molecular Medicine, Faculty of Medicine, University of Malaya. 50603 Kuala Lumpur, Malaysia.
2. Department of Biomedical Engineering, Faculty of Engineering, University of Malaya. 50603 Kuala Lumpur, Malaysia.
3. Department of Pharmacology, Faculty of Medicine, University of Malaya, 50603 Kuala Lumpur, Malaysia.
4. Department of Chemistry, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia.
Received 2014-2-20; Accepted 2014-7-1; Published 2014-8-1
Abstract
Platelet rich plasma clot- releasate (PRCR) shows significant influence on tissue regeneration in clinical trials. Although, the mechanism of PRCR effect on fibroblast differentiation has been studied on 2D culture system, a detailed investigation is needed to establish the role of PRCR in cell seeded in 3D scaffolds. Therefore, a study was conducted to evaluate the influence of PRCR in fibroblasts (DFB) differentiation and extracellular matrix formation on both 3D and 2D culture systems. Cell viability was measured using MTT assay and DFB differentiation was evaluated by determining the expression levels of nucleostamin and alpha smooth muscle actin (α-SMA), using indirect immunostaining and Western blotting. The expression levels of extracellular matrix genes (collagen-I, collagen-III, fibronectin and laminin) and focal adhesion formation gene (integrin beta-1) were measured using Real-time PCR. The PRCR at 10% showed significant effect on cells viability compared with 5% and 20% in both culture environments. The decrease in the expression levels of nucleostamin and the increase in α-SMA signify the DFB differentiation to myofibroblast-like cells that was prominently greater in 3D compared to 2D culture. In 3D culture systems, the total collage production, expression levels of the extracellular matrix gene and the focal adhesion gene were increased significantly compared to 2D culture. In conclusion, 3D culture environments enhances the proliferative and differentiation effects of PRCR on DFB, thereby potentially increases the efficacy of DFB for future tissue engineering clinical application.
Keywords: Platelet rich plasma, human skin fibroblast, growth factors, PCL-CA scaffold.
Introduction
Wound healing is a complex process in reparation of damaged tissues through inflammatory, proliferative and remodeling events. The consecutive events of wound healing are initiated and regulated by some of essential growth factors that are secreted by different types of cells [1]. Blood platelets have been considered as a main source of growth factors which arrive firstly to the position of the damaged tissue and release their granular contents of growth factors into the wound milieu [2, 3]. Most of the growth factors secreted by activated blood platelets are involved in wound healing processes including, transforming growth factors beta (TGF- β), epidermal growth factors (EGF), fibroblast growth factors (FGF), vascular endothelial growth factors (VEGF) and granulocyte macrophage colony stimulating factors (GM-CSF) [4, 5]. Previous studies reported significant outcome in wound repair process after applying exogenous form of growth factors that showed significant decrease in the formation of scar tissue [6]. However, PDGF-BB is the only growth factor approved by the Food and Drug Administration (FDA) [7]. In addition, the daily application of the exogenous growth factor could be considered as a disadvantage compared with the practical use of PRCR. Furthermore, there are some reconsiderations about potential problems of applying exogenous growth factors must be made to use them clinically in acute or chronic wounds [8, 9].
Platelets rich plasma clot-releasate (PRCR) is a natural reservoir of growth factors which represents an alternative approach for successful wound healing in cost- and time- effective manner. The PRCR has also been referred to as platelet-enriched plasma, platelet-rich concentrate, autologous platelet gel, and platelet releasate [10]. It is derived from the non-coagulated blood and contains approximately five times of platelets more than the baseline of whole blood platelets account [11, 12]. Therefore, the activated PRCR is able to release high levels of the essential growth factors which stimulate progenitor cells proliferation, migration, differentiation and subsequently wound matrix formation [13].
In skin wound healing, dermal fibroblasts play an important role in the healing process through its differentiation to myofibroblasts [1]. The contractile activity of myofibroblasts and their ability in producing extracellular matrix proteins are considered as a main factor in skin wound healing [14, 15]. However, there is a limited information of the possible role of PRCR in inducing skin fibroblast differentiation to myfibroblast and the secretion of the extracellular matrix in 2D and 3D scaffold culture environments. The outcome of the current study indicates that PRCR potentially stimulate skin fibroblast differentiation to myofibroblast with higher efficacy in 3D compared to 2D culture systems as measured by the expression levels of the differentiation markers. Furthermore, the extracellular proteins that have an important role in wound healing were observed to be highly up regulated in 3D compared to 2D culture environments.
Methods
Skin fibroblast and PRCR preparation
Adult human skin fibroblast cells were purchased from Lonza (Lonza, USA, cat. # CC-2511) and expended in growth medium consisting of Dulbecco's modified Eagle's medium (DMEM; Gibco -Invitrogen, USA) supplemented with 20% fetal bovine serum (FBS; Gibco -Invitrogen, USA), 100 μM 2-mercaptoethanol (Sigma-Aldrich, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco -Invitrogen, USA) and incubated at standard conditions (humidified atmosphere, 5% CO2, 37°C) for enough expansion. The blood (10 ml) was collected in EDTA-tube from 5 healthy volunteers and centrifuged at 400 g for 15 min and the supernatant that contained platelet rich plasma (PRR) was collected in a new tube. Then, the PRR were incubated with 25 mM CaCl2 at room temperature for 1 hour in order to activate the platelets. The activated PRR was then centrifuged at 2500g for 15 min to collect soluble releasate of PRCR that was used in the following experiments. The concentrations of TGF-β1 in whole blood and PRCR preparations were measured using an ELISA kit (Human TGF-β1 Quantikine ELISA Kit, R&D system, Cat. # DB100B). In total, 7 samples of blood samples or PRCR were used to obtain TGF-β1 measurements.
Polymer scaffold preparation
High purity citric acid (CA), polycaprolactone triol (PCLT; Mw = 300) and dioxane were purchased from Sigma-Aldrich and used as received. The polymer synthesis was conducted as follows: equimolar amounts of both the CA and PCL were mixed together and the reactant mixture was heated up to 160-165 ºC until CA crystals melted. The reaction mixture was further mixed at 140-145 ºC for 1 h under a constant stream of nitrogen. Thus formed polycaprolactone triol-citrate (PCL-CA) pre-polymer was processed into scaffolds by solvent-casting/particulate leaching method [16]. In brief, pre-polymer was dissolved in dioxane (20 % (w/w) solution) and the solution was mixed with sieved sodium chloride (NaCl) crystals (sieve size: 200-300 µm). The polymer-salt-solvent slurry was placed in TeflonTM molds and cured in oven at 70-80°C for 7 days. After that, the solid polymer-salt composite was taken out of the mold and placed in water for salt leaching. The PCLT-CA scaffolds were freeze-dried prior to use.
Seeding of skin fibroblast into PCLT-CA scaffolds
Before seeding the cells into scaffold, the scaffold was placed in 24 well plates, washed 3 times with PBS, left to dry at room temperature and placed under UV for overnight. The sterilised scaffold was soaked with FBS-free DMEM for 3 h, the media were removed and the scaffold was left for another 3 h in the incubator. Then, the desired amount of the cells were dropped into the scaffold and left for another 1 h in the incubator (37°C and 5% CO2) before top up with the media.
Cell Proliferation Assay
The PRCR at 5% and 10% percent was diluted in DMEM growth media. The fibroblast cells were seeded at 1x104 cells per well in triplicate at optimal conditions (37˚C, 5% CO2 in humidified incubator) in 96 well plates incubated for 48 h. A MTT assay was used to determine the in vitro cells viability. DMEM media containing 5% and 10% FBS were used as controls DMEM media containing 20% DMSO was used as a negative control. Following 48 h incubation, a 10 μl solution of freshly prepared 5 mg/ml MTT in PBS was added to each well and allowed to incubate for an additional 2 to 4 h. Then, the media was removed and DMSO was added at 100 μl/well. Plates were then swirled gently to facilitate formazan crystal solubilization. The absorbance was measured at 570 nm using a microplate reader (Tecan Infinite M200 Pro). The percent of cell viability was calculated as follows: 100 - (Absorbance PRCR-treated cells/Absorbance untreated cells) × 100.
Indirect Immunostaining
Cells were grown in 2D culture (5×105/T75 flask) or seeded into 3D culture system (5×105/scaffold) and treated with 10% PRCR for 7 days. Then, cells were trypsinised and reseeded on cover slides fixed in 6-well plates and for 24 h. Next, the cells were washed three times with PBS to remove the PRCR residues and fixed with ice-cold methanol for 15 min at -20ºC. After the washing steps, the cells were incubated with a coating buffer for 1 h at room temperature. A mouse antibody specific to alpha smooth muscle actin (abcam, USA, cat. # ab7817), nucleostamin (Santa Cruz, USA, cat. # sc-166460), collage I (abcam, USA, cat. # ab90395), collagen III (abcam, USA, cat. # ab3610), Intergin β1 (Santa Cruz, USA, cat. # sc-374429) was add separately and the cells were incubated for overnight at 4ºC. The cells were washed three times with PBS and incubated for 30 min with an anti-mouse IgG labeled with FITC fluorescent dye (Invitrogen, USA, cat. # 62-6511). To stain the cell nuclei, Hoechst dye was added (Invitrogen, USA, cat. # H1399) for the last 15 min of the incubation period. To determine nucleostemin positive cells, equal views for each sample were selected to count approximately 250 cells for each group of treatment. Then, the percentage of nucleostemin-positive cells was calculated by dividing the number of nucleostemin-positive cells by total number of cells sampled.
Western blotting
Western blot analysis was performed as previously described [17]. In brief, cells lysates were prepared for immunoblotting using ice-cold lyses buffer. The amount of protein in cell lysates was quantified to ensure equal loading (20 μg) in Western blot gels using 2-D Quant Kit (GE Healthcare Bio-Sciences, USA) according to the manufacturer's instructions. The separated proteins were transferred onto nitrocellulose membrane and then blocked with blocking buffer. The membrane was incubated overnight with antibody specific to the target proteins as mentioned above or anti-GAPDH antibody (abcam, USA, cat. # ab8245) as a loading control protein. The membrane was then washed again with Tris Buffered Saline as described previously, and finally developed using Western Blue®stabilized substrate (Promega, USA). Relative quantification of protein markers compared to untreated control were analyzed using Image J software.
Scanning electron microscopy (SEM)
PRCR-treated fibroblasts were cultured for 3 days into 3D culture system (1×105/scaffold) and then fixed with 3% glutaraldehyde in PBS for 24 h at 4°C. After thorough washing with PBS, scaffold samples were dehydrated sequentially in 50%, 70%, 95% and 100% ethanol. Then the fixed samples were freeze-dried, sputter coated with gold and observed under SEM (HitachiS-530).
Cellular Production of Total Collagen
Skin fibroblasts were seeded into T25 flask (5×105/T75 flask, n=6) or into scaffolds (5×105/scaffold, n=6) and grown in maintenance medium (2% FBS as control), 5% PRCR and 10% PRCR for 7 days. After the cell-conditioned medium was collected, cells were detached by trypsinisation. To measure total soluble collagen, total soluble collagen kit was used (QuickZyme BioSciences, Leiden-Netherlands). Briefly, the cells were trypsinised in both culture systems, washed with PBS and equal number of cells were used for this assay. The PBS was replace by 0.5 M Acetic acid (250 μl/well of 24-well-plate), incubate overnight at 4oC on a rotating platform, The cellular extract was centrifuged 10 minutes 3.000 x g, and the supernatant was tested in the assay in 1-fold to 10-fold dilutions made in dilution buffer. Measurements were performed at a wavelength of 540 nm in triplicates using Tecan Infinite M200 spectrophotometer (Tecan Group Ltd., Switzerland). A standard curve for calculating collagen concentration was obtained using a manufacturer-supplied acid soluble type I collagen calibration standard solution.
Real time PCR
Real time PCR was used to study the expression level of extracellular gene markers collagen I, collagen III, fibronectin, laminin, integrin β1 and GAPDH as an endogenous gens as previously described [17]. In brief, skin fibroblasts were plated at 106/ T75 flask or scaffold and maintained in FBS free DMEM medium supplemented with 10% (Treatment group) or 10% FBS (Control group). Cells were harvested after 72 h and total RNA was extracted using Trizol method (Invitrogen, USA), purified by RNA purification kit (Promega, USA) and quantified by nanospectrophotometer. The amount of 1 µg of pure RNA was used for gene expression analysis. Reverse-transcription was performed using cDNA synthesis kit (Invitrogen, USA) to prepare first strand cDNA. Real time-PCR was performed using SYBR Green PCR kit (BioRad, USA). The PCR program consisted of an initial step of 10 sec at 95°C, followed by 40 cycles of denaturing at 95°C, annealing at 50°C for 5 second and extension at 60°C for 31 sec. The relative level of genes expression was quantified using GAPDH gene as an endogenous control.
Statistical analysis
All the assays were done in triplicates and the statistical analyses were performed using GraphPad Prism version 5.01(GraphPad Software, San Diego, CA). P values of <0.05 were considered significant. Error bars are expressed as ± SD.
Results
The effects of PRCR on fibroblasts morphology and cell viability
This study was initiated by evaluating the effects of PRCR on skin fibroblasts morphology and viability. PRCR is rich with growth factors especially TGF-β1 that showed high concentration (3.3 ± 0.2 ng/ml) compared to the whole blood (0.05 ± 0.01 ng/ml) as presented in Figure 1A. Then, cells viability was evaluated by MTT assay that showed significant (p<0.001) increase in the viability of the cells treated with 5% and 10% compared to 20% of PRCR in both 2D and 3D culture environment (Fig. 1B). Cell viability of PRCR-treated cells in 3D culture was significantly higher than 2D culture and the cells in both culture systems showed lessen viability at high percentage of PRCR. Therefore, we tested the efficacy of the PRCR on cell function in the following experiments at 10% that showed the higher cell viability. Considerable morphological changes were observed in PRCR- treated fibroblasts compared to FBS-treated cells. After 14 days of treatment, the PRCR induced elongated spindle shape of cells that also appeared to be separated individually at the low density cell culture (Fig. 1C). This observation is in contrast with irregular shapes of control fibroblasts (Fig. 1D). The SEM images show the pore size of scaffold (Fig. 1E), pore distribution (Fig. 1F) and the surface morphology of pore walls (Fig. 1G). After 3 days in culture, the confluent fibroblasts and the extracellular matrix covered the pore surface area of the scaffold and the cells appeared to be embedded into the extracellular matrix (Fig. 1H).
Cells viability and morphology after the treatment with PRCR: (A) TGF-β1 levels in PRCR and whole blood (WB); (B) Cell viability of PRCR-treated fibroblast was measured by MTT assay in 2D and 3D culture systems calculated as a percentage of viability; (C) skin fibroblast after 14 days of treatment with 10% PRCR possess single and elongated spindle shape compared to untreated cells; (D) fibroblast treated with 10% FBS as control; (E) scaffold with diameter 0.5 cm used as a 3D culture system; (F and G) SEM images of PCL-CA non-seeded scaffolds (bars: 1 mm and 100 µm for E and F respectively); (H) PCL-CA scaffold with dermal fibroblast cells treated with 10% PRCR after 3 days in culture (Bar: 50 µm).

The effects of PRCR on fibroblast differentiation to myofibroblast
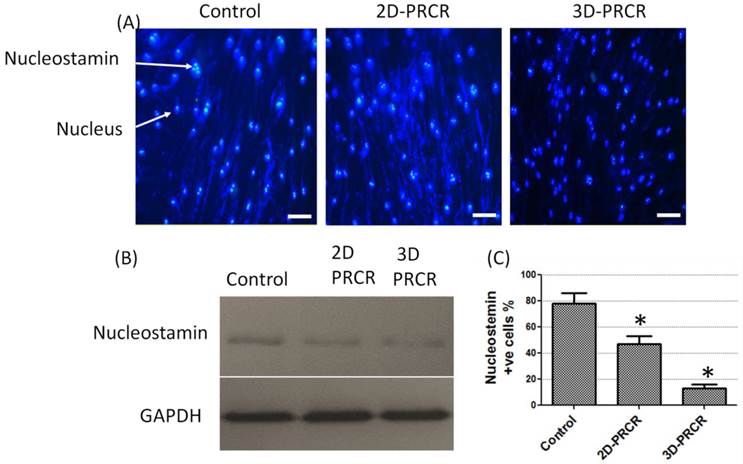
The morphological changes after PRCR treatment indicated that the fibroblasts had undergone a differentiation process into myofibroblast-like cells. To confirm this observation, further analysis was performed using indirect immunostaining (Fig. 2A) and Western blotting analysis (Fig. 2B). Nucleostamin, a positive marker of non-differentiated cells like fibroblast, was first analysed in PRCR-treated fibroblasts. The results showed that the treated fibroblasts with 10% of PRCR exhibit lower level of nucleostamin expression compared to control. In addition, the fibroblasts that were seeded in PCL-CA scaffolds showed lower expression level of nucleostamin compared to 2D culture. The semi-quantitative analysis showed that approximately 80% of cells in control culture were nucleostemin-positive, whereas the number was decreased to less than 50% for PRCR treatment cells in 2D culture and less than 15% in 3D culture system (Fig. 2C).
Nucleastamin immunostaining test: (A) indirect immunostaining of nucleostamin in PRCR-treated fibroblast cultured in 2D system (Control and 2D-PRCR) or seeded in the scaffold (3D system), treated with 10% PRCR for 7 days, trypsinised and reseeded on coverslip for 24 h; the immunostaining was applied using anti-nucleostamin antibody and secondary antibody labeled with FITC fluorescence dye (bar = 20 µm); (B) Western blotting of PRCR-treated cells using anti-nucleostamin antibody; (C) nucleostamin positive cells (*P<0.001, One-Way ANOVA).
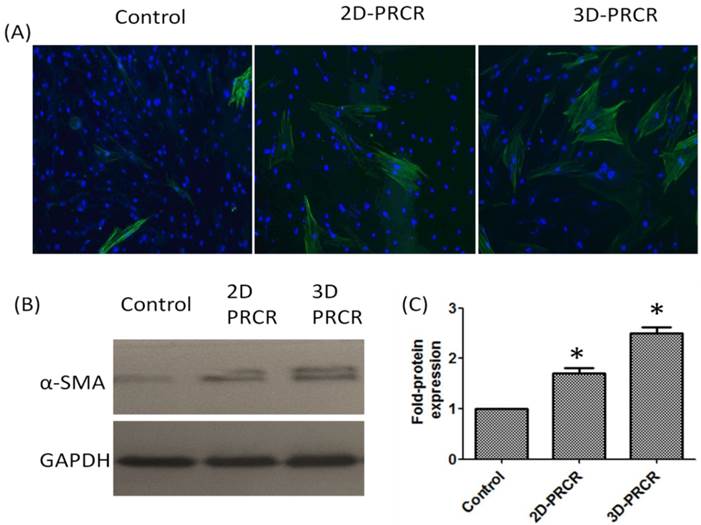
To characterise the types of cells after fibroblast differentiation as a result of PRCR treatment, we evaluated the expression level of alpha smooth muscle actin (α-SMA), a positive marker of myofibroblast that was highly expressed in the PRCR-treated fibroblast compared to control (FBS-treated cells) as represented in Figure 3A. PRCR-treated fibroblast in both culture systems (2D and 3D) showed higher expression level of α-SMA protein compared to control. However, cells seeded in 3D expressed α-SMA protein at higher levels after the treatment with PRCR compared with 2D culture (Fig. 3B). The analysis of Western blot image by Image J software showed that the expression level of α-SMA protein in PRCR-treated fibroblast was significantly (P<0.001) higher than the expression level in control and 3D culture showed 2.5 ± 0.1 fold higher in α-SMA protein expression compared with control while 2D culture showed 1.7 ± 0.1 fold compared with control (Fig. 3C).
The expression level of alpha smooth muscle actin (α-SAM) protein: (A) cells were grown in flask (2D culture system) or seeded into scaffold (3D culture system) and treated with 10% PRCR for 7 days; cells were trypsinised and reseeded on cover slides for 24 h and fixed with ice-cold methanol for immunostaining; the indirect immunostaining was carried out using anti- α-SAM antibody and secondary antibody labeled with FITC fluorescence dye; (B) Western blotting of PRCR-treated cells using anti- α-SAM antibody; (C) the fold of α-SAM expression using image J software analysis (*P<0.001, One-Way ANOVA).
Effects of PRCR on collagen production in 2D and 3D culture conditions
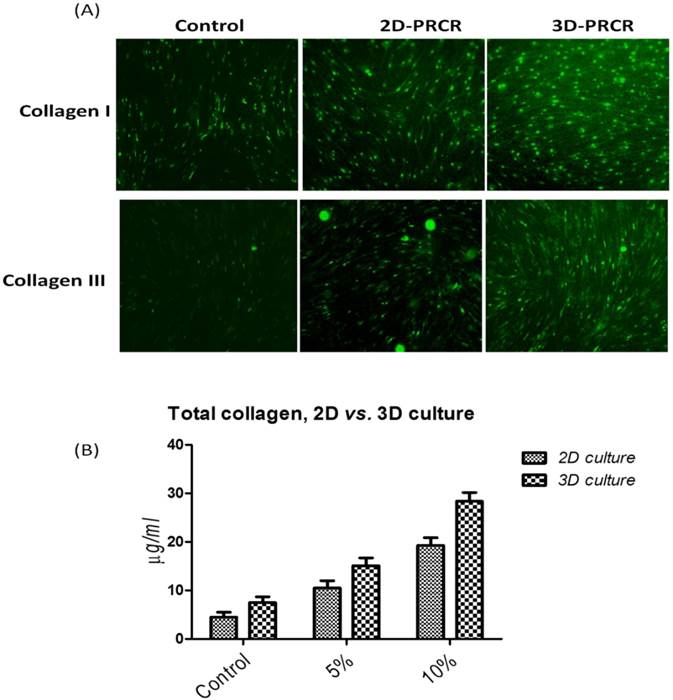
Skin fibroblasts were cultured in 2D and 3D culture systems to study the effects of PRCR treatment on extracellular matrix formation. The cells were seeded into flask or scaffold and treated with PRCR for 7 days or FBS as controls. Then, the cells were reseeded on cover slip for immunostaining or used directly to determine total cellular soluble collagen. PRCR treatment increased the expression levels of collagen types I and III, as evidenced by indirect immunostaining using anti- collagen I and anti- collagen III antibodies (Fig. 4). The results showed that total collagen production by these activated fibroblast was significantly (P<0.001) increased compared with control cells under the same culture conditions. In addition, the effects of PRCR treatment on the production of total collage increased with increasing PRCR dosage (Fig. 5C). Interestingly, the 3D culture condition showed significant (P<0.01, Two-way ANOVA) ability to induce collage production compared with the 2D culture (Fig. 5D). The difference in the amount of total collagen between control groups of 2D (4.5 ± 1.1 µg/ml) and 3D (7.5 ± 1.3 µg/ml) was insignificant (P>0.05 Two-way ANOVA,). However, at the doses of 5% and 10% of PRCR, the 3D culture showed higher production of total collagen compared to the 2D culture. For example, at the dose of 5% PRCR, the production of total collage was 10.5 ± 1.5 µg/ml of 2D culture compared with 15.1 ± 1.6 µg/ml of 3D culture. While, higher difference was observed at the dose of 10% PRCR (19.3 ± 1.6 µg/ml of 2D culture vs. 28.4 ± 1.8 µg/ml of 3D culture).
The expression levels of collagen I and collagen III and total collagen production after the treatment with PRCR in 2D and 3D culture environment; (A) skin fibroblasts in 2D and 3D culture systems were treated with 10% PRCR for 7 days, reseeded on cover slides for 24 h and fixed with ice-cold methanol for immunostaining; the indirect immunostaining was carried out using anti-collagen I and collagen III antibodies and secondary antibody labeled with FITC fluorescence dye; (B) cellular production of total collagen after the treatment with 5% and 10% PRCR in 2D and 3D culture systems.
Study the expression level of extracellular gene markers collagen I, collagen III, fibronectin and laminin using Real time- PCR. Skin fibroblast were plated at 106/ T75 flask (2D culture) or scaffold (3D culture) and maintained in FBS free DMEM medium supplemented with 10% PRCR (Treatment group) or 10% FBS (Control group). Cells were harvested after 72 h and total RNA was extracted and proceeded for RT-PCR (the amount of 1 µg of pure RNA was used for gene expression analysis). PRCR treatment increased the expression level of collagen I and collagen III compared with control in 2D and 3D cultures. The results also showed the effect of the environment on the expression level of collagen I (P<0.05, One-way ANOVA) and collagen III (P>0.05, One-way ANOVA) in 2D compared to 3D culture. Significant increase in the expression level of fibronectin and laminin in the PRCR-treated cells compared with control in the 2D (P<0.05, One-way ANOVA) and 3D environment (P<0.01, One-way ANOVA).
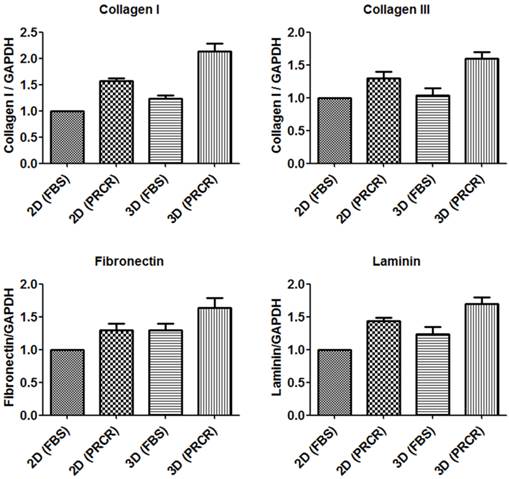
Gene expression analysis of extracellular matrix genes
Total RNA was extracted from both 2D culture and PCL-CA scaffolds, purified and precisely quantified to use for gene expression analysis by Real-time PCR. Gene expression of major extracellular proteins collagen I, collagen III, fibronectin and laminin were evaluated after the treatment with PRCR and normalized to the expression level of GAPDH as an endogenous gene. The results showed that the PRCR treatment increased the expression level of collagen I (1.5 ± 0.1) and collagen III (1.3 ± 0.2) compared with FBS-treated cells in 2D culture. Similarly, in the 3D culture environment, the expression level of collagen I (2.2 ± 0.3) and collagen III (1.6 ± 0.2) of PRCR-treated fibroblast were higher than the expression levels of these genes of FBS-treated cells as represent in Figure 6. Furthermore, the results also showed the effect of the environment on the expression level of collagen I (P<0.05, One-way ANOVA) and collagen III (P>0.05, One-way ANOVA) in 2D compared to 3D culture. The expression level of fibronectin and laminin increased after the PRCR treatment compare with untreated cells. The results showed significant (P<0.05, One-way ANOVA) increase in the expression level of fibronectin (1.3 ± 0.2) and laminin (1.4 ± 0.1) in the PRCR-treated cells compared with FBS-tread cells in the 2D environment. Likewise, the expression level of fibronectin (1.6 ± 0.3) and laminin (1.7 ± 0.2) at 3D environment was also increased after the treatment with PRCR compared to FBS-treated cells cultured in 3D system (1.3 ± 0.2 and 1.2 ± 0.2 respectively). Moreover, the 3D culture environment showed significant (P<0.05) increase in the expression level of fibronectin and laminin compared with 2D culture environment.
The expression level of integrin β1, a cell membrane protein of focal adhesion formation: (A) Indirect immunostainning showed that the treatment with PRCR increased the expression of integrin β1 in human skin fibroblast; (B) real-time PCR of integrin β1 (P<0.01, One-way ANOVA).
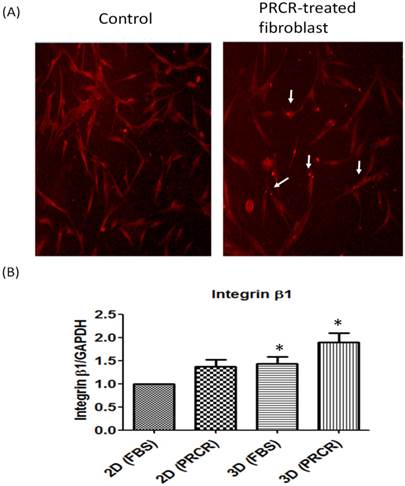
Interestingly, the significant increase in the expression level of extracellular matrix genes was associated with an increase in the expression level of integrin β1, a cell membrane protein that forms focal adhesion regions with the extracellular matrix. PRCR-treated fibroblast in 3D culture system showed the highest expression of integrin β1 (1.9 ± 0.4) compared with control cells (1.4 ± 0.3) in 3D (P<0.05) and control cells in 2D culture systems (P<0.01).
Discussion
Dermal fibroblasts present an important role in wound healing through successive proliferation, differentiation and extracellular matrix formation [1]. It has been known that cell responses to 3D matrices (scaffolds) differ from cells grown on 2D surfaces [18-20]. Therefore, it is reasonable to consider that dermal fibroblast would most likely respond to PRCR stimulation at different levels based on culture systems. Thus, we conducted this study in order to evaluate whether PRCR treatment may modulate dermal fibroblast responses in porous scaffolds at higher extent than 2D culture systems. The results of this study showed that the PRCR induced fibroblast differentiation to myofibroblast-like cells and the response of skin fibroblast to PRCR effect in 3D environment was greater than 2D culture in terms of cell viability, differentiation, and extracellular matrix formation.
Since it is a known fact that activated blood platelets produce important growth factors in wound healing cascade, in the reported study the PRCR treatment considerably improved cell viability and induced the fibroblasts differentiation into myofibroblasts. As described previously, this phenomenon is evidenced by decrease nucleostamin expression, a stemness protein marker, and increase the expression level of α-SMA which is a myofibroblast protein maker. As anticipated, PRCR-treated dermal fibroblasts showed higher responses in cell-seeded scaffolds compared to 2D culture system. The data of this study has shown considerable increase in cell viability as measured by MTT assay in addition to a considerable decrease in nucleostamin expression and increase in the expression level of α-SMA as evaluated by immunostaining and Western blotting analysis. Previous studies showed that the mesenchymal stem cells (MSC) exhibited higher level of chondrogenic marker expression after the PRCR treatment of cells inside the scaffold environment compared to the 2D culture [21]. Even mature chondrocyte when was seeded in the 3D systems showed high viability and efficacy in the proliferation and expression of chondrogenic gene markers after the treatment with PRCR [22]. Another study showed opposite results, as the treatment of the ligament fibroblasts with high doses of PRCR in the scaffold culture system showed negative responses in terms of cells function. The reported study also indicated to the positive effect of the low doses of PRCR in 3D culture environment [23]. In our study, the optimization of PRCR has been done by using the cell proliferation assay (MTT) in order to evaluate the cell viability after the treatment with PRCR. In 3D scaffold culture, the 10% PRCR in culture medium demonstrated higher cell viability compared to 5% PRCR that was used in the following experiments.
Determination of the optimal dose of PRCR leads to definition of the appropriate levels of the main growth factors that are responsible for cells proliferation and maturation. For example, TGF-β1, a growth factor detected at high concentrations in PRCR, has been identified as an important inducer of the myofibroblastic phenotype [24-26]. Human skin fibroblasts generally show significant expression of myofibroblast marker (α-SMA) after the treatment with PRCR. In terms of wound healing process, the main characteristic of myofibroblasts is the high expression of α-SMA that transform into actin stress fibers in order to gain the required contractile activity for efficient wound closure [14, 15]. Furthermore, myofibrblasts have been described as an activated fibroblasts that are able to produce extracellular matrix proteins based on the overlapped steps of wound healing process [27].
A well-known fact is that the cell adhesion and migration, modulated by growth factors, are essential processes in wound healing and tissue regeneration [28]. The extracellular matrix formation plays the key role in tissue formation. From our experiments presented in this paper the activated skin fibroblast produced higher quantity of collagen I and collagen III in 3D scaffold compared to 2D culture as evaluated by immunosatining and total collagen production. In addition, gene expression analysis by Real time- PCR showed higher expression level in 3D compared to 2D culture of extracellular matrix genes of collagen I, collagen III, fibronectin and laminin. Furthermore, the results of this study also showed higher expression of cell membrane adhesion protein, intergrin β1 in 3D compared to 2D culture. It has been known that the activated fibroblast during the wound healing trends to move from surrounding collagenous connective tissues and to attach to a new fibronectin-fibrin extracellular matrix and subsequently synthesize its collagenous matrix [29, 30]. Furthermore the up regulation of integrin β1 after the treatment with PRCR is important for cell attachment to the extracellular matrix. Integrins are the molecules primarily responsible for cell adherence to extracellular matrix, and expression of these molecular components is modulated actively by different growth factors [31]. PRCR are abundant with the essential growth factors such as TGF-β1 and EGF that up regulate β1- and β3-integrin, and PDGF-BB that induces a5-integrin receptors [32, 33]. In addition, the adhesion of activated fibroblast to extracellular matrix is mainly stimulated by physiological concentrations of PRCR-growth factors [34-36]. In conclusion, we found in this study that the 3D culture environment of porous PCL-CA synthetic scaffolds provide a convenient microenvironment conditions to the PRCR- activated fibroblast to proliferate and synthesis a new extracellular matrix as evidenced by the higher expression level of the collagen, fibronectin and laminin compared to the 2D culture.
Acknowledgements
This project was funded by the University of Malaya and Ministry of Science, Technology and Innovation (ERGS grant ER016-2013A and UMRG grant RG309-14AFR).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci. 2004;9:283-289
2. Mehta V. Platelet-Rich Plasma: A Review of the Science and Possible Clinical Applications. Orthopedics. 2010;33:2. doi: 10.3928/01477447-20100104-22
3. Andia I, Maffulli N. Platelet-rich plasma for managing pain and inflammation in osteoarthritis. Nature. 2013;9:721-730 doi:10.1038/nrrheum.2013.141
4. Behm B, Babilas P. et al. Cytokines, chemokines and growth factors in wound healing. J Eur Acad Dermatol Venereol. 2012;26(7):812-820
5. Amable PR, Carias RB, Teixeira MV, da Cruz Pacheco I, Corrêa do Amaral RJ, Granjeiro JM, Borojevic R. Platelet-rich plasma preparation for regenerative medicine: optimization and quantification of cytokines and growth factors. Stem Cell Res Ther. 2013;4(3):67
6. Gope R. The effect of epidermal growth factor & platelet-derived growth factors on wound healing process. Indian J Med Res. 2002;116:201-206
7. Goldman R. Growth factors and chronic wound healing: past, present, and future. Adv Skin Wound Care. 2004;17(1):24-35
8. Fu X, Cheng B. et al. Growth factors and wound healing: review and prospect in recent ten years. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2004;18(6):508-512
9. Fu X, Li X. et al. Engineered growth factors and cutaneous wound healing: success and possible questions in the past 10 years. Wound Repair Regen. 2005;13(2):122-130
10. Mehta S, Watson JT. Platelet rich concentrate: basic science and current clinical applications. J Orthop Trauma. 2008;22(6):432-8
11. Marx RE. Platelet-rich plasma (PRCR): What is PRCR and what is not PRCR? Implant Dent. 2001;10:225-228
12. Foster TE, Puskas BL, Mandelbaum BR. et al. Platelet-rich plasma: From basic science to clinical applications. Am J Sports Med. 2009;37:2259-2272
13. Demidova-Rice TN, Hamblin MR. et al. Acute and impaired wound healing: pathophysiology and current methods for drug delivery, part 2: role of growth factors in normal and pathological wound healing: therapeutic potential and methods of delivery. Adv Skin Wound Care. 2012;25(8):349-370
14. Hinz B, Mastrangelo B, Iselin CE, Chapponier C, Gabbiani G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am J Pathol. 2001;159:1009-1020
15. Katoh K, Kano Y, Amano M, Onishi H, Kaibuchi K, Fujiwara K. Rho-kinase-mediated contraction of isolated stress fibers. J Cell Biol. 2001;153:569-584
16. Djordjevic I. et al. Synthesis and characterization of novel citric acid-based polyester elastomers. Polymer. 2009(50):1682-1691
17. Rothan HA, Suhaeb AM, Kamarul T. Recombinant Human Adiponectin As A Potential Protein For Treating Diabetic Tendinopathy Promotes Tenocyte Progenitor Cells Proliferation and Tenogenic Differentiation In Vitro. International Journal of Medical Sciences. 2013;10(13):1899-1906 doi: 10.7150/ijms.6774
18. Cukierman E, Pankov R et al. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708-1712
19. Tamariz E, Grinnell F. Modulation of fibroblast morphology and adhesion during collagen matrix remodeling. Mol Biol Cell. 2002;13:3915-3929
20. Wenk MB. et al. Tenascin-C suppresses Rho activation. J Cell Biol. 2002;150:913-920
21. Drengk A. et al. Influence of platelet-rich plasma on chondrogenic differentiation and proliferation of chondrocytes and mesenchymal stem cells. Cells Tissues Organs. 2009;189(5):317-326
22. Lee HR. et al. Platelet-rich plasma loaded in situ-formed hydrogel enhances hyaline cartilage regeneration by CB1 upregulation. J Biomed Mater Res A. 2012;100(11):3099-3107
23. Yoshida R. et al. Increasing platelet concentration in platelet-rich plasma inhibits anterior cruciate ligament cell function in three-dimensional culture. J Orthop Res. 2014;32(2):291-5 doi: 10.1002/jor.22493
24. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349-363
25. Okuda K, Kawase T, Momose M. Platelet-rich plasma contains high levels of platelet-derived growth factor and transforming growth factor-b and modulates the proliferation of periodontally related cells in vitro. J Periodontol. 2003;74:849-857
26. Christgau M, Moder D, Hiller K-A, Dada A, Schmitz G, Schmalz G. Growth factors and cytokines in autologous platelet concentrate and their correlation to periodontal regeneration outcomes. J Clin Periodontol. 2006;33:837-845
27. Zhang K, Rekhter MC, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis: a combined immunohistochemical and in situ hybridization study. Am J Pathol. 1994;145:114-125
28. Xu J, Clark RAF. Extracellular matrix alters PDGF regulation of fibroblast integrins. J Cell Biol. 1996;132:239-249
29. Schaffer CJ, Nanney LB. Cell biology of wound healing. Int Rev Cytol. 1996;169:151-181
30. Gailit J, Clark RAF. Wound repair in the context of extracellular matrix. Curr Opin Cell Biol. 1994;6:717-725
31. Hynes RO. Integrins: Versatility, modulation and signalling in cell adhesion. Cell. 1992;69:11-25
32. Heino J, Massague J. Transforming growth factor-b switches the pattern of integrins expressed in MG-63 human osteosarcoma cells and causes a selective loss of cell adhesion to laminin. J Biol Chem. 1989;264:21806-21811
33. Bellas RE, Bendori R, Farmer SR. Epidermal growth factor activation of vinculin and b1-integrin gene expression in quiescent Swiss 3T3 cells: Regulation through a protein kinase C-independent pathway. J Biol Chem. 1991;266:12008-12014
34. Smith PC, Caceres M, Martinez J. Induction of the myofibroblastic phenotype in human gingival fibroblasts by transforming growth factor-b1: Role of RhoA-ROCK and c-Jun N-terminal kinase signalling pathway. J Periodontal Res. 2006;41:418-425
35. Zhang J, Wang JH. Platelet-rich plasma releasate promotes differentiation of tendon stem cells into active tenocytes. Am J Sports Med. 2010;38(12):2477-86 doi: 10.1177/0363546510376750
36. Zhang J, Middleton KK, Fu FH, Im H-J, Wang JH-C. HGF Mediates the Anti-inflammatory Effects of PRCR on Injured Tendons. PLoS ONE. 2013;8(6):e67303. doi:10.1371/journal.pone.0067303
Author contact
![]() Corresponding author: Hussin A. Rothan, Department of Molecular Medicine, Faculty of Medicine, University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail addresses rothanedu.my.
Corresponding author: Hussin A. Rothan, Department of Molecular Medicine, Faculty of Medicine, University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail addresses rothanedu.my.