Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(10):1015-1021. doi:10.7150/ijms.8330 This issue Cite
Research Paper
Rosiglitazone Enhances Apolipoprotein M (Apom) Expression in Rat's Liver
Guanghua Luo1, Yuehua Feng1, Jun Zhang1, Qinfeng Mu1, Yuanping Shi1, Li Qin1, Lu Zheng1, Maria Berggren-Söderlund3, Peter Nilsson-Ehle3, Xiaoying Zhang2 ![]() , Ning Xu3
, Ning Xu3 ![]()
1. Comprehensive Laboratory, the Third Affiliated Hospital of Soochow University, Changzhou 213003, P.R. China.
2. Department of Cardiothoracic Surgery, the Third Affiliated Hospital of Soochow University, Changzhou 213003, P.R. China;
3. Division of Clinical Chemistry and Pharmacology, Department of Laboratory Medicine, Lunds University, S-221 85 Lund, Sweden.
Received 2013-12-10; Accepted 2014-7-14; Published 2014-7-29
Abstract
Apolipoprotein M (APOM) has been suggested as a vasculoprotective constituent of high density lipoprotein (HDL), which plays a crucial role behind the mechanism of HDL-mediated anti-atherosclerosis. Previous studies demonstrated that insulin resistance could associate with decreased APOM expressions. In agreement with our previous reports, here, we further confirmed that the insulin sensitivity was also reduced in rats treated with high concentrations of glucose; such effect could be reversed by administration of rosiglitazone, a peroxisome proliferator-activated receptor-γ (PPARγ). The present study shows that Apom expression is significantly affected by either rosiglitazone or hyperglycemia alone without cross interaction with each other, which indicates that the pathway of Apom expression regulating by hyperglycemia might be differed from that by rosiglitazone. Further study indicated that hyperglycemia could significantly inhibit mRNA levels of Lxrb (P=0.0002), small heterodimer partner 1 (Shp1) (P<0.0001), liver receptor homologue-1 (Lrh1) (P=0.0012), ATP-binding cassette transporter 1 (Abca1) (P=0.0012) and Pparb/d (P=0.0043). Two-way ANOVA analysis demonstrated that the interactions between rosiglitazone and infusion of 25% glucose solution on Shp1 (P=0.0054) and Abca1 (4E, P=0.0004) mRNA expression was statistically significant. It is concluded that rosiglitazone could increase Apom expression, of which the detailed mechanism needs to be further investigated. The downregulation of Apom by hyperglycemia might be mainly through decreasing expression of Pparg and followed by inhibiting Lxrb in rats.
Keywords: Apolipoprotein M, Hyperglycemia, Insulin sensitivity, Liver receptor homologue-1, ATP-binding cassette transporter 1.
Introduction
Human apolipoprotein M (APOM) is mainly found in hepatocytes of the liver and tubular epithelial cells in kidney [1], and it could also been expressed weakly in the colorectal tissues [2]. The physiological roles of APOM were explored gradually during recent years. It has recently been documented that APOM can protect endothelium by delivering sphingosine-1-phosphate (S1P) to the S1P1 receptor [3] and enhance the antioxidant effect of high density lipoprotein (HDL), in which the most abundant protein is APOA-I [4]. Also, it has been demonstrated that APOM is important for the formation of preβ-HDL and cholesterol efflux to HDL, which could attenuate atherosclerotic process [5].
We have previously reported that hepatic APOM levels were significantly decreased in hyperglycemic rats, and in cell cultures high concentrations of glucose inhibited APOM expression [6]. It was previously demonstrated that hyperglycemia could increase the flux of free fatty acids (FFAs) by accelerating lipolysis, which may induce or aggravate insulin resistance in the liver and muscle through altering the insulin signaling pathway [7, 8]. In light of evidence that mice with genetic defects of leptin (ob/ob mouse), exhibiting resistance to insulin, also showed significantly lower levels of Apom [9], we speculate that down-regulation of APOM expression by hyperglycemia may be associated with insulin resistance.
Rosiglitazone, a peroxisome proliferator-activated receptor-γ (PPARγ) agonist used for the treatment of type 2 diabetes, is a potent insulin sensitizer that promotes glucose uptake by adipose tissue, skeletal muscle and liver [10-12]. Apart from improving insulin resistance, recent evidences demonstrated that rosiglitazone has multieffects, including attenuating airway inflammation by inhibiting the proliferation of effector T cells [13], reducing hippocampal neuronal damage [14], improving endothelial function [15], increasing cell surface GLUT4 level [16], preventing pulmonary fibrosis [17] and even preventing graft-versus-host disease [18]. However, it remains unknown whether rosiglitazone affects APOM in vivo. In the present study we examined the effects of rosiglitazone and hyperglycemia on Apom expression in a rat's models. According to our previous investigation showing that liver X receptor (LXR) [19, 20] and ATP-binding cassette transporter A1 (ABCA1) [21] could involve in the regulation of APOM expression. In the present study, we further examined the effects of rosiglitazone and hyperglycemia on expression of Pparb/d and genes of LXR signaling pathway including its target gene, Abca1 in rat's models in order to find possible mechanism(s) of Apom regulation by rosiglitazone and hyperglycemia.
Materials and methods
Animal experiments. The experimental protocols were approved by the Animal Care and Use Committee of Soochow University, Suzhou, China. In general, each experimental group contained 6-8 adult male Sprague-Dawley rats (250-300g). Rats were commercially obtained from the Shanghai Slac Laboratory Animal Co., China or the Changzhou Cavens Laboratory Animal Co., China. Rats were kept in separate cages in a temperature-controlled (22°C) room with 12-hrs light-dark cycle and were provided with standard rodent chow and water ad libitum. The rats were acclimatized for one week before the experiments.
In the present study, all rats were pre-operated and two small catheters were placed in a jugular vein seven days before the liquid infusion. In brief, rats were anesthetized with 10% chloral hydrate (4ml/kg). Two catheters were placed in a jugular vein, one for infusion of 25% glucose solution (10ml·kg-1·h-1 for 6 h, HuaYu Pharmaceutical Co.) or insulin solution (10mU·kg-1·min-1 during HEC (hyperinsulinemic euglycemic clamp), Wanbang Biopharmaceuticals) and another for infusion of 20% glucose solution (during HEC). An additional catheter was placed in a carotid artery for blood sampling (also seven days before the experiments). The free ends of both catheters were attached to long segments of steel tubing and tunneled subcutaneously on the back of the neck. The catheters were flushed with isotonic saline containing 50 IU/ml heparin (Qianhong Bio-pharma Co.) and filled with a viscous solution of heparin (500 IU/ml) and 300 g/L polyvinyl pyrolidone (PVP-10; Sigma) to prevent refluxing of blood into the catheter lumen.
When performing liquid infusions, the catheters were carefully connected to the infusion pumps (Smiths Medical). 25% glucose solution or insulin /20% glucose solution (during HEC) was infused for determining gene expressions or for assessing insulin sensitivity, respectively. Additionally, liquid was infused via tail vein in two parallel experiments which were performed to avoid the interference of the HEC test on gene expression. All the control rats were given 5% glucose solution. In the studies of rosiglitazone and/or infusion of 25% glucose solution on gene expressions, the rats were dosed daily with 0.25% (w/v) sodium carboxymethycellulose (CMC, Sinopharm Chemical Reagent Co., Ltd) or rosiglitazone (Higher Biotech Co., Ltd, 4mg·kg-1, containing 0.25% CMC) via oral gavage for 4-5 days. The HEC technique was applied for assessing insulin sensitivity in rats (n=8 for each group) that were gavaged with 0.25% (w/v) CMC or rosiglitazone (dissolved in 0.25% CMC, at a concentration of 4mg·kg-1·d-1) for 4 days, then infused with either 5% or 25% glucose solution intravenously for 5hrs. In another set of experiments without assessment of insulin sensitivity, rats (n=6) were gavaged with 0.25% (w/v) CMC or rosiglitazone (dissolved in 0.25% CMC, with the concentration of 4mg·kg-1·d-1) for 5 days, then infused with either 5% or 25% glucose intravenously for 6hrs. After glucose infusion, blood samples and liver tissues were collected for plasma FFAs and Apom mRNA levels detection, respectively.
Reverse transcription and real-time RT-PCR. Total RNA in rats' tissues was extracted according to the manufacturer's instructions using a total RNA purification kit (Omega Bio-Tek). The quality of the RNA samples was determined by the absorbance measurements at 260/280 nm. Using the first strand cDNA synthetic kit (Qiagen) according to the manufacturer's instructions, 2μg total RNA was reverse transcribed to cDNA. The mRNA levels of the target and reference genes were measured under real-time PCR using TaqMan technology. The PCR primer sets were designed according to the information of GenBank, as listed in Table 1. B-actin was used as reference genes. Relative standard curves were produced to compensate for the efficiency of the PCRs. Quantification of target genes mRNA levels was relative to B-actin mRNA level. The real-time PCR reaction for each gene was performed in a 25μL volume, in a glass capillary containing 0.1μL 100μM each primer and probe, 2μL cDNA, 2.5μL 10xbuffer, 1.5μL MgCl2 (25mM), 0.5μL dNTP (10mmol/L), and Taq DNA polymerase 0.5μL. Thermal cycling conditions included the following steps: initial denaturation at 95 ºC for 3 min, followed by 40 cycles at 95 ºC for 5 sec and 60 ºC for 15 sec (rat Lxra, 62 ºC for 15 sec; rat Apom, 58ºC for 12 sec; rat B-actin, 61ºC for 10 sec). All PCRs were performed on the LightCycler (Roche, Switzerland) real-time PCR system.
Sequences of primers and probes.
| Gene | Primer/Probe | Sequence (5' to 3') |
|---|---|---|
| Rat Apom | Forward primer | acaaagagaccccagagccc |
| Reverse primer | tccatggtgggagccg | |
| Probe | FAM-acctgggcctgtggtactttattgctgg-TAMRA | |
| Rat B-actin | Forward primer | gccactgccgcatcctct |
| Reverse primer | ctggaagagagcctcgggg | |
| Probe | FAM-agctgcctgacggtcaggtcatcactatc-TAMRA | |
| Rat Lxra | Forward primer | ggagcacgctacatttgccata |
| Reverse primer | cctcttcttgacgcttcagtttctt | |
| Probe | FAM-tggccactgccccatggacaccta-TAMRA | |
| Rat Lxrb | Forward primer | tgtcggggcagcggaac |
| Reverse primer | gctcctcagaaagcacgcac | |
| Probe | FAM-atggatgccttcatgcggcgca-TAMRA | |
| Rat Shp1 | Forward primer | cttccctttgaccacagccg |
| Reverse primer | ctgactggcgatgtaggtcttagag | |
| Probe | FAM-acatcccagggtctgattacatcaatgc-TAMRA | |
| Rat Lrh1 | Forward primer | tgtcctgtgtgtggcgataaag |
| Reverse primer | ggactgttcgcttaaagaaaccc | |
| Probe | FAM-tctgggtatcattacggtctcctcacctgt-TAMRA | |
| Rat Abca1 | Forward primer | gagaccaaccaggcaatccag |
| Reverse primer | ttgatgagcgtgacttcggtt | |
| Probe | FAM-cgatatctcgattcatggagtgtgtcaacc-TAMRA | |
| Rat Pparb/d | Forward primer | gaagaaccgcaacaagtgtcagta |
| Reverse primer | ccttccaaagcggatagcgt | |
| Probe | FAM-cttccagaagtgcctggcgctcggc-TAMRA |
Determinations of FFAs. FFAs were determined with the nonesterified fatty acid (NEFA) colorimetric method (Applygen Technologies Inc, Beijing, China). Briefly, total plasma FFAs were extracted with chloroform: N-heptane: methanol (56:42:2), coupled with copper, reacted with color reagent and measured with a colorimeter at 550 nm. The standard curve was produced using a series of dilution of palmitic acid.
Statistics. Data are expressed as means ± SEM. Statistical analyses were performed with the GraphPad Prism 6.0 software (GraphPad Software, Inc., San Diego, California, USA). Multiple comparisons were performed with one-way ANOVA, and comparisons between two groups were statistically evaluated by the unpaired t-test. Cross interaction was analyzed by two-way ANOVA. P-values less than 0.05 were considered significant.
Results
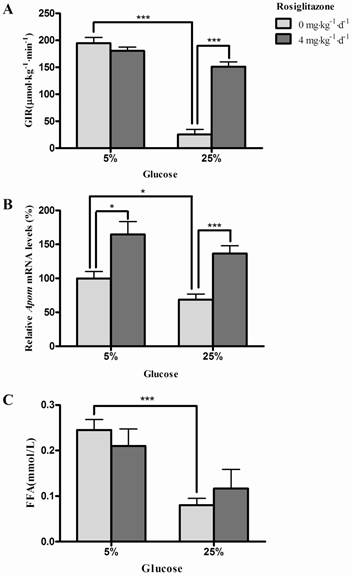
Effects of glucose and/or rosiglitazone on insulin sensitivity, hepatic Apom mRNA levels and plasma FFAs in rats
Both 25% glucose solution (P<0.0001) and rosiglitazone (P<0.0001) affect the glucose infusion rates (GIR) (Fig. 1A). Furthermore, the marked inhibition of GIR by 25% glucose could be reversed by administration of rosiglitazone (P<0.001) (Fig. 1A). Fig. 1B shows that hepatic Apom mRNA levels in rats were significantly decreased after infusion of 25% glucose solution (P=0.0356). Rosiglitazone significantly increased the hepatic Apom mRNA levels in groups with 5% (P=0.0132) or 25% (P=0.0007) glucose administration. However, the interaction of rosiglitazone and glucose on Apom mRNA expression was not statistically significant (P=0.8981). As shown in Fig. 1C, the plasma FFAs were clearly lower in rats infused with 25% glucose solution than in rats infused with 5% glucose solution (P=0.0005), and rosiglitazone had no significant effect on plasma FFA levels in these rats (P=0.9790). Two-way ANOVA indicated that there was no interaction between rosiglitazone and glucose on plasma levels of FFAs (P=0.2656).
Effects of glucose and rosiglitazone on insulin sensitivity, hepatic Apom mRNA levels and plasma FFAs. (A) GIR during hyperinsulinemic euglycemic clamp. (B) Hepatic Apom mRNA levels. (C) Effects of glucose and rosiglitazone on plasma FFAs. Data are presented as mean ± SEM. *P <0.05 and ***P<0.001 vs. control.
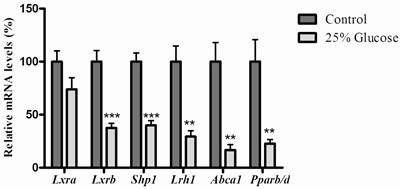
Effects of glucose and rosiglitazone on expression of genes of LXR signaling pathway, Abca1 and Pparb/d mRNA expression in rat liver
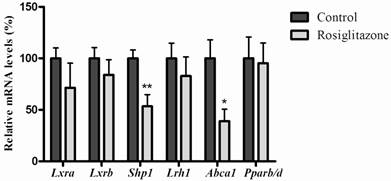
As shown in figure 2, when rats infused with 25% glucose solution, hepatic mRNA levels of Lxrb were significantly inhibited (P=0.0002), and small heterodimer partner 1 (Shp1) (P<0.0001), liver receptor homologue-1 (Lrh1) (P=0.0012), ATP-binding cassette transporter 1 (Abca1) (P=0.0012) and Pparb/d (P=0.0043) were also significantly reduced, while rosiglitazone only decreased mRNA levels of Shp1 (P=0.0074) and Abca1 (P=0.0171) (Fig. 3).
Effects of glucose on genes related to the LXR signaling pathway and mRNA level of Pparb/d in rat liver. Rats were infused with 5% glucose solution (controls, n=6) or 25% glucose solution (n=6) intravenously for 6hrs. Data are presented as means ± SEM. **P <0.01 and ***P<0.001 vs. controls.
Effects of rosiglitazone on genes related to the LXR signaling pathway and mRNA level of Pparb/d in rats' liver. Rats (n=6 for each group) were gavaged with 0.25% (w/v) CMC (Controls) or rosiglitazone (dissolved in 0.25% CMC, at a concentration of 4mg·kg-1·d-1) for 5 days. Data are presented as means ± SEM. *P <0.05 and **P <0.01 vs. controls.
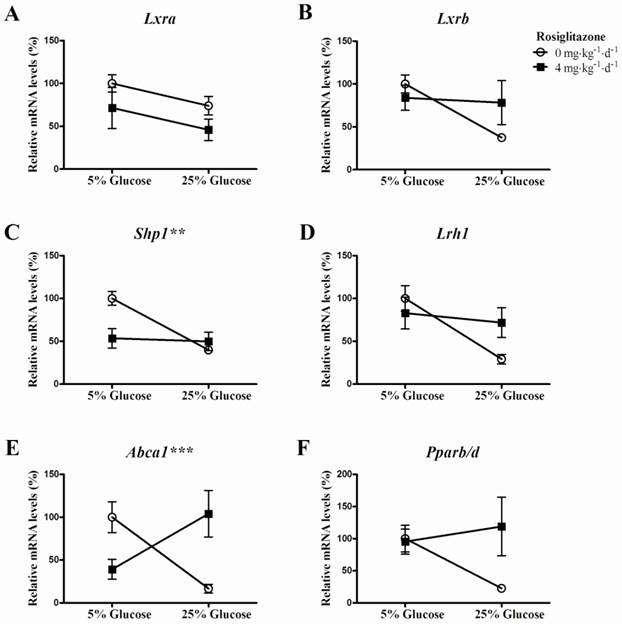
Cross interactions between rosiglitazone and infusion of 25% glucose solution on genes of LXR signaling pathway, Abca1 and Pparb/d mRNA expression
Two-way ANOVA analysis showed that the interactions between rosiglitazone and infusion of 25% glucose solution on Shp1 (Fig. 4C, P=0.0054) and Abca1 (Fig. 4E, P=0.0004) mRNA expression was statistically significant. And, the interactions on Lxra (Fig. 4A, P=0.9844), Lxrb (Fig. 4B, P=0.0861), Lrh1 (Fig. 4D, P=0.0592) and Pparb/d (Fig. 4F, P=0.0759) were not statistically significant.
Cross interaction between rosiglitazone and glucose on target genes of LXR signaling pathway and Pparb/d mRNA expression. Rats (n=6 for each group) were gavaged with 0.25% (w/v) CMC or rosiglitazone (dissolved in 0.25% CMC, at a concentration of 4mg·kg-1·d-1) for 5 days, then infused with 5% glucose solution (controls) or 25% glucose solution intravenously for 6 hrs. Data are presented as means ± SEM. **P <0.01 and ***P <0.001 vs. controls.
Discussion
In the present study, the rats in the control group were administrated with 5% glucose solution, to avoid the starving on normal physiological metabolic processes.
In agreement with previous reports [22-26], the insulin sensitivity was reduced in rats treated with high concentrations of glucose and this effect could be reversed by rosiglitazone. The present study shows that Apom expression is also significantly affected by either rosiglitazone or hyperglycemia alone without cross interaction with each other, which suggests that the pathway of Apom expression regulated by hyperglycemia might be differed from that by rosiglitazone. Rosiglitazone is well documented, it acts through the PPARγ pathway and alleviates insulin resistance by reducing uptake of free fatty acids as well as enhancing lipometabolism [27]. However, in the present study, plasma FFA levels did not increase as expected, but rather decreased plasma FFAs occurred when rats were infused with 25% glucose solution. It is possible that hyperglycemia can elicit insulin secretion [28] and insulin therefore suppress the fatty acid release through inhibiting lipolysis [29] in this experimental model. We previously reported that activation of neither PPARα nor PPARγ influenced APOM expression in HepG2 cells [30]. While interestingly, our present data demonstrated that activation of PPARγ by the rosiglitazone could up-regulate hepatic Apom expression in rats, which suggests that the signal pathway of PPARγ on regulation of Apom expression might be much more complicated in vivo than in vitro, or perhaps, the difference between rat Apom gene and human APOM gene contributes to the regulation patterns of PPARγ.
ABCA1 is an important member of the ATP-binding cassette-transporter family and is involved in the apoAI-mediated cholesterol efflux from macrophages [31]. PPARγ activator i.e., rosiglitazone and troglitazone, could enhance ABCA1 expression in primary human monocyte-derived macrophages [32], whereas ciglitazone had no effect in cultured human keratinocytes [33]. In this study, hepatic Abca1 mRNA expression was down-regulated by either rosiglitazone or 25% glucose, but rosiglitazone could totally reverse the hyperglycemia-induced down-regulation of Abca1 expression, which demonstrated that down-regulation of Abca1 expression induced by hyperglycemia, might be mediated via the PPARγ pathway in rats.
Our previous study indicated that upregulation of ABCA1 could elevate APOM expression. So Abca1 down-regulated by rosiglitazone should suppress Apom in rat accordingly, but in present study, rosiglitazone significantly increased Apom expression, which suggested that upregulation of Apom induced by rosiglitazone might not be regulated through Abca1, which is possible that rosiglitazone or PPARγ could bind to Apom promoter region directly regulating its expression. More detailed mechanism needs further investigation.
To explore the mechanism of down-regulation of Apom by hyperglycemia, we analyzed mRNA expressions of genes related to the pathway of liver X receptor. Infusion of 25% glucose solution significantly decreased hepatic mRNA levels of Lxrb, Shp1, Lrh1, Abca1 and Pparb/d in rats. We previously reported that palmitic acid-induced upregulation of PPARB/D expression could significantly inhibit APOM expression in HepG2 cells [34]. So by inference, inhibition of Pparb/d mediated by infusion of 25% glucose solution could elevate mRNA levels of Apom. However, the final result showed that infusion of 25% glucose solution significantly decreased Apom expression in rats. One reasonable explanation is that hyperglycemia might suppress Apom expression through multiple signal pathways in vivo. Venteclef, et al., have demonstrated that LRH1 could directly regulate human and mouse Apom transcription by binding to the LRH1 response element located in the proximal APOM promoter region, and bile acids suppressed APOM expression by inhibiting LRH1 transcriptional activity [35]. We have demonstrated that activation of LXR triggers upregulation of ABCA1 and subsequently increases APOM expression [21]. The results of present study showed that downregulation of genes (Lxrb, Shp1, Lrh1) of LXR signaling pathway and its target genes, Abca1 by hyperglycemia could be inhibited by rosiglitazone, although the interactions between rosiglitazone and hyperglycemia on Lxrb (Fig. 4B, P=0.0861) and Lrh1 (Fig. 4D, P=0.0592) did not reach the statistically significant levels. We therefore speculated that hyperglycemia suppresses Apom expression mainly via decreasing expression of Pparg and followed by inhibiting Lxrb in rat.
Acknowledgements
This research project was supported by the National Natural Science Foundation of China (NSFC) (81071414), the Natural Science Foundation of Jiangsu Province (BK2011245), a research grant from the Changzhou Science & Technology Bureau (CJ20122012) and Jiangsu Provincial 333 High-level Talents Cultivation Project (BRA2013062).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang XY, Dong X, Zheng L, Luo GH, Liu YH, Ekstrom U. et al. Specific tissue expression and cellular localization of human apolipoprotein M as determined by in situ hybridization. Acta histochemica. 2003;105:67-72
2. Luo G, Zhang X, Mu Q, Chen L, Zheng L, Wei J. et al. Expression and localization of apolipoprotein M in human colorectal tissues. Lipids in health and disease. 2010;9:102
3. Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M. et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A. 2011;108:9613-8
4. Elsoe S, Ahnstrom J, Christoffersen C, Hoofnagle AN, Plomgaard P, Heinecke JW. et al. Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis. 2012;221:91-7
5. Wolfrum C, Poy MN, Stoffel M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nature medicine. 2005;11:418-22
6. Zhang X, Jiang B, Luo G, Nilsson-Ehle P, Xu N. Hyperglycemia down-regulates apolipoprotein M expression in vivo and in vitro. Biochimica et biophysica acta. 2007;1771:879-82
7. Green A, Rumberger JM, Stuart CA, Ruhoff MS. Stimulation of lipolysis by tumor necrosis factor-alpha in 3T3-L1 adipocytes is glucose dependent: implications for long-term regulation of lipolysis. Diabetes. 2004;53:74-81
8. Delarue J, Magnan C. Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care. 2007;10:142-8
9. Xu N, Nilsson-Ehle P, Hurtig M, Ahren B. Both leptin and leptin-receptor are essential for apolipoprotein M expression in vivo. Biochemical and biophysical research communications. 2004;321:916-21
10. Le Bouter S, Rodriguez M, Guigal-Stephan N, Courtade-Gaiani S, Xuereb L, de Montrion C. et al. Coordinate Transcriptomic and Metabolomic Effects of the Insulin Sensitizer Rosiglitazone on Fundamental Metabolic Pathways in Liver, Soleus Muscle, and Adipose Tissue in Diabetic db/db Mice. PPAR Res. 2010;2010:1-17
11. Lebovitz HE, Dole JF, Patwardhan R, Rappaport EB, Freed MI. Rosiglitazone monotherapy is effective in patients with type 2 diabetes. J Clin Endocrinol Metab. 2001;86:280-8
12. Young PW, Cawthorne MA, Coyle PJ, Holder JC, Holman GD, Kozka IJ. et al. Repeat treatment of obese mice with BRL 49653, a new potent insulin sensitizer, enhances insulin action in white adipocytes. Association with increased insulin binding and cell-surface GLUT4 as measured by photoaffinity labeling. Diabetes. 1995;44:1087-92
13. Zhao Y, Huang Y, He J, Li C, Deng W, Ran X. et al. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma agonist, attenuates airway inflammation by inhibiting the proliferation of effector T cells in a murine model of neutrophilic asthma. Immunol Lett. 2014;157:9-15
14. Sayan-Ozacmak H, Ozacmak VH, Barut F, Jakubowska-Dogru E. Rosiglitazone treatment reduces hippocampal neuronal damage possibly through alleviating oxidative stress in chronic cerebral hypoperfusion. Neurochem Int. 2012;61:287-90
15. Gao XQ, Li HW, Ling X, Qiu YH, Gao Y, Zhang Y. Effect of rosiglitazone on rabbit model of myocardial ischemia-reperfusion injury. Asian Pac J Trop Med. 2013;6:228-31
16. Martinez L, Berenguer M, Bruce MC, Le Marchand-Brustel Y, Govers R. Rosiglitazone increases cell surface GLUT4 levels in 3T3-L1 adipocytes through an enhancement of endosomal recycling. Biochem Pharmacol. 2010;79:1300-9
17. Jin GY, Bok SM, Han YM, Chung MJ, Yoon KH, Kim SR. et al. Effectiveness of rosiglitazone on bleomycin-induced lung fibrosis: Assessed by micro-computed tomography and pathologic scores. Eur J Radiol. 2012;81:1901-6
18. Song EK, Yim JM, Yim JY, Song MY, Rho HW, Yim SK. et al. Rosiglitazone prevents graft-versus-host disease (GVHD). Transpl Immunol. 2012;27:128-37
19. Zhang X, Zhu Z, Luo G, Zheng L, Nilsson-Ehle P, Xu N. Liver X receptor agonist downregulates hepatic apoM expression in vivo and in vitro. Biochemical and biophysical research communications. 2008;371:114-7
20. Zhu C, Di D, Zhang X, Luo G, Wang Z, Wei J. et al. TO901317 regulating apolipoprotein M expression mediates via the farnesoid X receptor pathway in Caco-2 cells. Lipids in health and disease. 2011;10:199
21. Di D, Wang Z, Liu Y, Luo G, Shi Y, Berggren-Soderlund M. et al. ABCA1 upregulating apolipoproein M expression mediates via the RXR/LXR pathway in HepG2 cells. Biochemical and biophysical research communications. 2012;421:152-6
22. Oakes ND, Kennedy CJ, Jenkins AB, Laybutt DR, Chisholm DJ, Kraegen EW. A new antidiabetic agent, BRL 49653, reduces lipid availability and improves insulin action and glucoregulation in the rat. Diabetes. 1994;43:1203-10
23. Eldershaw TP, Rattigan S, Cawthorne MA, Buckingham RE, Colquhoun EQ, Clark MG. Treatment with the thiazolidinedione (BRL 49653) decreases insulin resistance in obese Zucker hindlimb. Horm Metab Res. 1995;27:169-72
24. Kramer D, Shapiro R, Adler A, Bush E, Rondinone CM. Insulin-sensitizing effect of rosiglitazone (BRL-49653) by regulation of glucose transporters in muscle and fat of Zucker rats. Metabolism. 2001;50:1294-300
25. Zhao Z, Lee YJ, Kim SK, Kim HJ, Shim WS, Ahn CW. et al. Rosiglitazone and fenofibrate improve insulin sensitivity of pre-diabetic OLETF rats by reducing malonyl-CoA levels in the liver and skeletal muscle. Life Sci. 2009;84:688-95
26. Gao W, Jusko WJ. Modeling disease progression and rosiglitazone intervention in type 2 diabetic Goto-Kakizaki rats. J Pharmacol Exp Ther. 2012;341:617-25
27. Song GY, Gao Y, Wang C, Hu SG, Wang J, Qu DM. et al. Rosiglitazone reduces fatty acid translocase and increases AMPK in skeletal muscle in aged rats: a possible mechanism to prevent high-fat-induced insulin resistance. Chin Med J (Engl). 2010;123:2384-91
28. Chirieac DV, Chirieac LR, Corsetti JP, Cianci J, Sparks CE, Sparks JD. Glucose-stimulated insulin secretion suppresses hepatic triglyceride-rich lipoprotein and apoB production. Am J Physiol Endocrinol Metab. 2000;279:E1003-11
29. Boden G, Chen X, Desantis RA, Kendrick Z. Effects of insulin on fatty acid reesterification in healthy subjects. Diabetes. 1993;42:1588-93
30. Xu N, Ahren B, Jiang J, Nilsson-Ehle P. Down-regulation of apolipoprotein M expression is mediated by phosphatidylinositol 3-kinase in HepG2 cells. Biochimica et biophysica acta. 2006;1761:256-60
31. Lawn RM, Wade DP, Garvin MR, Wang X, Schwartz K, Porter JG. et al. The Tangier disease gene product ABC1 controls the cellular apolipoprotein-mediated lipid removal pathway. J Clin Invest. 1999;104:R25-31
32. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP. et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nature medicine. 2001;7:53-8
33. Jiang YJ, Lu B, Kim P, Elias PM, Feingold KR. Regulation of ABCA1 expression in human keratinocytes and murine epidermis. Journal of lipid research. 2006;47:2248-58
34. Luo G, Shi Y, Zhang J, Mu Q, Qin L, Zheng L. et al. Palmitic acid suppresses apolipoprotein M gene expression via the pathway of PPARbeta/delta in HepG2 cells. Biochemical and biophysical research communications. 2014;445:203-7
35. Venteclef N, Haroniti A, Tousaint JJ, Talianidis I, Delerive P. Regulation of anti-atherogenic apolipoprotein M gene expression by the orphan nuclear receptor LRH-1. The Journal of biological chemistry. 2008;283:3694-701
Author contact
![]() Corresponding author: Xiaoying Zhang MD., Department of Cardiothoracic Surgery, the Third Affiliated Hospital of Soochow University, Changzhou 213003, P.R. China. Tel: +86 519 68871278; Fax: + 86 519 86621235; E-mail: zhangxy6689996com and Ning Xu, MD, PhD., Division of Clinical Chemistry and Pharmacology, Department of Laboratory Medicine, Lunds University, Lund S-221 85, Sweden. Tel: +46 46 173487; Fax: + 46 46 130064; E-mail: ning.xulu.se.
Corresponding author: Xiaoying Zhang MD., Department of Cardiothoracic Surgery, the Third Affiliated Hospital of Soochow University, Changzhou 213003, P.R. China. Tel: +86 519 68871278; Fax: + 86 519 86621235; E-mail: zhangxy6689996com and Ning Xu, MD, PhD., Division of Clinical Chemistry and Pharmacology, Department of Laboratory Medicine, Lunds University, Lund S-221 85, Sweden. Tel: +46 46 173487; Fax: + 46 46 130064; E-mail: ning.xulu.se.