Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Experimental procedures
Results and discussion
Conclusion
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(9):955-961. doi:10.7150/ijms.9316 This issue Cite
Research Paper
Cyclic Phosphatidic Acid Inhibits Alkyl-Glycerophosphate-Induced Downregulation of Histone Deacetylase 2 Expression and Suppresses the Inflammatory Response in Human Coronary Artery Endothelial Cells
Tamotsu Tsukahara1 ![]() , Hisao Haniu2, Yoshikazu Matsuda3
, Hisao Haniu2, Yoshikazu Matsuda3
1. Department of Hematology and Immunology, Kanazawa Medical University, 1-1 Daigaku, Uchinada, Ishikawa 920-0293, Japan
2. Institue for Biomedical Sciences, Shinshu University Interdisciplinary Cluster for Cutting Edge Research 3-1-1 Asahi, Matsumoto, Nagano 390-8621, Japan
3. Clinical Pharmacology Educational Center, Nihon Pharmaceutical University, Ina-machi, Saitama 362-0806, Japan
Received 2014-4-7; Accepted 2014-6-10; Published 2014-7-5
Abstract
Activation of the endothelium by alkyl-glycerophosphate (AGP) has been implicated in the development of atherosclerosis. Our previous study suggested that cyclic phosphatidic acid (cPA) inhibits arterial wall remodeling in a rat model in vivo. However, the mechanisms through which specific target genes are regulated during this process remain unclear. Here, we examined whether cPA inhibited AGP-induced expression of class I histone deacetylases (HDACs, namely HDAC1, HDAC2, HDAC3, and HDAC8), which may affect subsequent transcriptional activity of target genes. Our experimental results showed that human coronary artery endothelial cells (HCAECs) expressed high levels of HDAC2 and low levels HDAC1, HDAC3, and HDAC8. Moreover, AGP treatment induced downregulation of HDAC2 expression in HCAECs. However, cotreatment with cPA inhibited this downregulation of HDAC2 expression. Interestingly, treatment with AGP increased the expression and secretion of endogenous interleukin (IL)-6 and IL-8; however, this effect was inhibited when HCAECs were cotreated with cPA or the synthetic peroxisome proliferator-activator receptor gamma (PPARγ) antagonist T0070907. Thus, our data suggested that cPA may have beneficial effects in inflammation-related cardiovascular disease by controlling HDAC2 regulation.
Keywords: Histone deacetylase, coronary artery endothelial cells, cyclic phosphatidic acid
Introduction
Atherosclerosis is an inflammatory disease of the arterial wall. Increased alkyl-glycerophosphate (AGP) serum levels are an important risk factor for atherosclerosis, and AGP accumulation in the vessel wall has been suggested to trigger neointima formation [1, 2], an early step in the development of atherosclerotic plaques. Atherogenic lesions subsequently progress through a prolonged process of lipid accumulation caused by buildup of plaque and deposits of fatty substances in the interior walls of the arteries. However, some clinical studies have shown that neointima formation induced by unsaturated lysophosphatidic acid (LPA) is not mediated by the LPA G-protein-coupled receptors (GPCRs) LPA1 and LPA2, which are the major LPA receptor subtypes expressed in the vessel wall [1].
A previous report demonstrated that topical application of AGP species onto uninjured carotid arteries of rodents induces arterial wall remodeling in a peroxisome proliferator-activated receptor gamma (PPARγ)-dependent manner [1]. Moreover, a study from our laboratory showed that cyclic phosphatidic acid (cPA) negatively regulates PPARγ function by stabilizing the silencing mediator of retinoid and thyroid hormone receptors (SMRT)-PPARγ complex [3], implying that cPA may have applications in ameliorating arterial dysfunction. cPA is a naturally occurring phospholipid, similar in structure to AGP, and can be generated by phospholipase D2 (PLD2) [2]. Despite structural similarities, cPA and AGP have several unique functions. For example, cPA inhibits cell proliferation, whereas AGP stimulates it [4]. Thus, further studies are required to elucidate the differential mechanisms through which these lipids exert their functions.
Histone deacetylases (HDACs), which have been shown to activate PPARγ and enhance the expression of its target genes [5, 6], have well-known functions in the regulation of chromatin structure and gene transcription via interactions with nuclear receptor corepressors, such as SMRT and nuclear receptor corepressor (NcoR) [7]. HDAC3 has been reported to inhibit PPARγ and nuclear transcription factor-κB (NF-κB) [8], and HDAC3 inhibition restores PPARγ function in obesity [7]. Additionally, HDAC2-containing complexes have been implicated in the regulation of nuclear receptor-dependent gene transcription [9]. In mammals, HDAC2 is predominantly localized in the nucleus in a wide range of tissues and cell types [10].
The inflammatory process involves activation of a variety of signaling pathways and expression of many target genes, which may be mediated by PPARγ, HDACs, and others, as discussed above. Furthermore, many studies have demonstrated a link between inflammatory mechanisms and atherosclerotic lesions. Indeed, prospective epidemiological studies have found increased vascular risk in association with increased basal levels of inflammation-related cytokines, such as interleukin (IL)-6 [11, 12]. Because the activation of inflammatory gene expression involves a large number of coregulators acting at different steps in the inflammatory process, additional studies are needed to fully elucidate the specific signaling mechanisms involved in inflammation-related gene expression.
In this study, we investigated the involvement of HDAC expression and activity in human coronary artery endothelial cells (HCAECs) and examined the effects of AGP and cPA on HDAC expression in order to elucidate the signaling mechanisms mediated by these two lipids. Our data have implications in the pathogenesis of atherosclerosis.
Experimental procedures
Materials
Mouse monoclonal anti-β-actin antibodies (sc-47778) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). An HDAC antibody sampler kit (#9928) and anti-HDAC8 antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). cPA (18:1) was a gift from Dr. Kimiko Murakami-Murofushi (Ochanomizu University, Tokyo, Japan). AGP (18:1) was purchased from Avanti Polar Lipids (Alabaster, AL, USA). An HDAC colorimetric activity assay kit was purchased from BioVision (Milpitas, CA, USA).
Cells and cultures
Primary HCAECs (Lonza, Walkersville, MD, USA) were propagated in endothelial cell growth medium (EBM-2) containing 5% fetal bovine serum (FBS), manufacturer-recommend supplemental growth factors (EGM-2 BulletKit), antibiotics, and antimycotics. All assays were performed in cells between passages three and 12 at 60%-80% confluence and were repeated at least three times in duplicate or triplicate.
Western blotting
Cells were seeded into 6-well plates (Iwaki, Tokyo, Japan) at a density of 1 × 105 cells/well. After the indicated treatments, cells were lysed on ice for 30 min in cell lysis buffer (20 mM Tris-HCl [pH 7.4], 10% [v/v] glycerol, 100 mM NaCl, 1% [v/v] Triton X-100, 1/100 protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA), and 1 mM dithiothreitol) and centrifuged at 16,000 × g for 20 min at 4°C. The supernatants were collected as cell lysates and assayed for protein content via the Bradford method using a Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Cell lysates were then separated on 5%-20% sodium dodecyl sulfate (SDS)-polyacrylamide gels (e-PAGEL; ATTO, Tokyo, Japan) and electrotransferred to Immobilon-P membranes (Millipore, Billerica, MA, USA). The membranes were blocked by Block Ace (DS Parma Biomedical Co. Ltd., Osaka, Japan) for 1 h and incubated with primary antibodies in Tris-buffered saline containing Tween 20 (TBS-T) with 5% Block Ace for 12 h at 4°C. Bands were visualized with EzWestLumi plus (ATTO).
Measurement of cell proliferation
HCAECs were seeded into 96-well plates at a density of 1 × 104 cells/well, and HDAC2 was knocked down using small-interfering RNA (siRNA), as described below. Cell proliferation was determined using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Ten microliters of Cell Counting Kit-8 solution was added to the medium and incubated for 2 h in an incubator with 5% CO2. The amount of orange formazan dye produced was calculated by measuring the absorbance at 450 nm in a microplate reader (Awareness Technology, Inc., Palm City, FL, USA).
Quantitative real-time reverse transcription polymerase chain reaction (RT-qPCR)
Total RNA was prepared from HCAECs using NucleoSpin RNA II (Takara, Ohtsu, Japan), and 0.05 µg of total RNA was then used for synthesis of cDNA using a ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan) according to the manufacturer's recommendations. Quantification of mRNA levels was carried out using an ECO Real-Time PCR system (Illumina, Inc., San Diego, CA, USA) and SYBR Green Realtime PCR Master Mix -Plus- (Toyobo). The following primer pair sets were used in this study: HDAC1, 5′-CTACTACGACGGGGATGTTGG-3′ (F) and 5′-GAGTCATGCGGATTCGGTGAG-3′ (R); HDAC2, 5′-ATGGCGTACAGTCAAGGAGG-3′ (F) and 5′-TGCGGATTCTATGAGGCTTCA-3′ (R); HDAC3, 5′-GCAAGGCTTCACCAAGAGTCT-3′ (F) and 5′-AGATGCGCCTGTGTAACGC-3′ (R); HDAC8, 5′-TCGCTGGTCCCGGTTTATATC-3′ (F) and 5′-TACTGGCCCGTTTGGGGAT-3′ (R); IL-6, 5′-CCTGAACCTTCCAAAGATGGC-3′ (F) and 5′-TTCACCAGGCAAGTCTCCTCA-3′ (R); and IL-8, 5′-ACTGAGAGTGATTGAGAGTGGAC-3′ (F) and 5′-AACCCTCTGCACCCAGTTTTC-3′ (R). A human housekeeping gene primer set (Takara) was used to provide optimal internal control/housekeeping genes according to the manufacturer's instructions. All PCR assays were performed in a 10-μL volume using 48-well PCR plates (Illumina, Hayward, CA, USA). The cycling conditions were as follows: 95°C for 10 min (polymerase activation), followed by 40 cycles of 95°C for 15 s, 55°C for 15 s, and 72°C for 30 s. Relative mRNA expression was calculated by using the arithmetic formula 2-ΔΔCq, where ΔCq is the difference between the threshold cycle of a given target cDNA and an endogenous reference cDNA.
siRNA
For knockdown of HDAC2 expression, HCAECs were transfected with siRNA targeting HDAC2 (sc-29345, Santa Cruz Biotechnology) using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA). Cells were seeded in 6-well plates at a density of 5 × 104 cells per well in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS. Cells were transfected with 100 pmol/mL of mRNA-specific siRNA or scrambled control siRNA. The reduction in HDAC2 levels was confirmed by western blot analysis.
HDAC activity assay
Measurement of HDAC activity in a cell-free system was performed using a colorimetric assay kit (VioBision, Milpitas, CA, USA). HeLa nuclear extracts (5 mg/mL) were incubated with or without LPA or cPA, and the reaction was initiated by the addition of HDAC substrate. After 30 min, the reaction was then terminated, and samples were read in an enzyme-linked immunosorbent assay (ELISA) plate reader (Awareness Technology, Inc., Palm City, FL, USA) at 405 nm.
IL-6 and IL-8 assays
The concentrations of IL-6 and IL-8 in the culture medium were determined using ELISA kits from Abcam (Cambridge, MA, USA) according to the manufacturer's instructions. LPA or cPA were dissolved in dimethyl sulfoxide (DMSO) to make a 10 mM stock solution that was stored at -80°C until use. HCAECs (5 × 104 cells/well) were incubated with or without LPA or cPA, and the cell culture media were centrifuged at 1,000 × g for 10 min to remove debris. Supernatants were collected, and the absorbance of each well was measured on a spectrophotometer (Awareness Technology) at 450 nm.
Statistical analysis
Student's t-tests were used for statistical comparisons. Differences were considered significant when the P-value was below 0.05.
Results and discussion
Comparison of endogenous class I HDAC expression in HCAECs
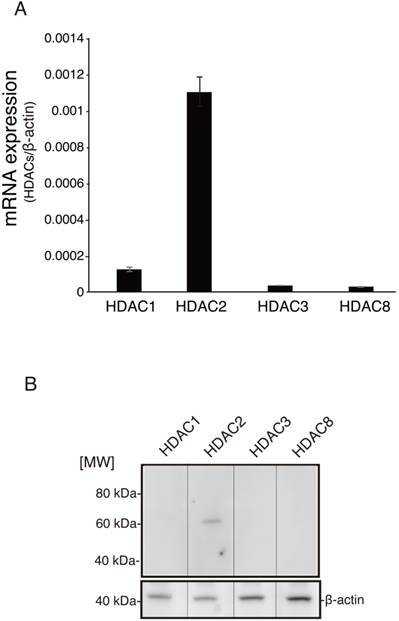
Four class I HDAC isoforms, i.e., HDAC1, HDAC2, HDAC3, and HDAC8, which have high sequence homology, have been shown to be expressed in humans [13]. Therefore, we analyzed the expression of class I HDACs in HCAECs. As shown in Fig. 1A, HDAC2 mRNA was expressed at high levels, while HDAC1, HDAC3, and HDAC8 were expressed at low levels. The expression of HDAC proteins, as measured by western blotting, confirmed these results (Fig. 1B). Interestingly, HDAC2 is a marker for heart- and muscle-related diseases [14] and plays an important role in cardiac stress responsiveness [15]. In addition, HDAC2-deficient mice develop cardiac hypertrophy [16]. Our results demonstrated that HDAC2 exhibited strong expression in HCAECs compared to other class I HDAC isoforms, therefore suggesting that alternative regulatory mechanisms mediate the expression of class I HDACs in human coronary arteries.
Comparison of endogenous class I HDAC expression in HCAECs. (A) Real-time PCR was used to measure HDAC1, HDAC2, HDAC3, and HDAC8 mRNA expression in HCAECs using specific primers for each HDAC isoform. The relative expression of each HDAC isoform was calculated by normalization to the expression of β-actin and was expressed as the mean ± SEM (n = 3). **p < 0.01. (B) Representative western blots showing HDAC1, HDAC2, HDAC3, and HDAC8 protein expression. HCAEC lysates (20 μg) were analyzed by SDS-PAGE and visualized with enhanced chemiluminescence reagent.
Effect of cPA on HDAC2 mRNA and protein expression in HCAEC
In previous studies, oxLDL has been shown to inhibit expression of HDAC1 and HDAC2 in endothelial cells [17], and HDAC2 has been shown to be downregulated in atherosclerotic lesions of human coronary arteries [17]. Therefore, we next investigated the mechanisms through which lysophospholipids mediate HDAC2 expression. As shown in Fig. 2A and B, HCAECs treated with 10 μM AGP exhibited reduced HDAC2 mRNA and protein expression by up to three-fold compared to vehicle alone. In contrast, cotreatment of HCAECs with cPA and AGP increased HDAC2 gene expression. Interestingly, combined treatment with AGP and the PPARγ antagonist T0070907 upregulated HDAC2 gene and protein expression, as compared with the AGP alone.
cPA modulated HDAC2 downregulation but was not an HDAC inhibitor. (A) Real-time PCR was used to measure the expression of HDAC2 mRNA in HCAECs treated with vehicle or the indicated PPARγ ligand for 20 h. The relative expression of HDAC2 was calculated by normalization to β-actin expression and was expressed as the mean ± SEM (n = 3). **p < 0.01. (B) Western blot analysis of HDAC2 protein expression in HCAECs treated with PPARγ ligand. HCAEC lysates (20 μg) were analyzed by SDS-PAGE and visualized with enhanced chemiluminescence reagent. (C) HCAECs were transfected with PPRE-Luc and CMV-β-galactosidase plasmids and treated with the indicated drugs (10 μM each) and control buffer (DMSO) for 20 h. Luciferase activity was measured in lysates of treated cells and normalized to β-galactosidase activity. Data are presented as the mean ± SEM (n = 3). **p < 0.01. (D) The effects of AGP and cPA on HDAC activity were examined by measuring the cleavage of acetylated histone H3 substrate. Data are presented as the mean ± SEM (n = 3). **p < 0.01.
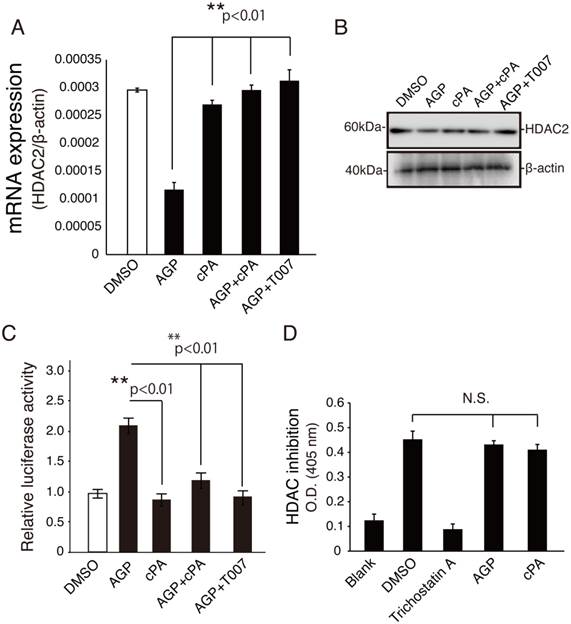
Next, to determine whether HCAECs expressed function PPARγ, we transfected HCAECs with a pGL3-PPRE-acyl-CoA oxidase luciferase reporter plasmid. After a 20-h treatment, the luciferase activity in HCAECs treated with 10 μM LPA was approximately 2.2-fold higher than that in vehicle (DMSO)-treated cells (Fig. 2C). As expected, combined treatment with cPA and T0070907 inhibited AGP-induced PPARγ activation. Moreover, cPA was able to modulate HDAC2 expression, but was not an HDAC inhibitor, as shown using a colorimetric assay (Fig. 2D). These results suggested that increased HDAC2 expression by cPA was mediated by inhibition of PPARγ. Because intimal hyperplasia due to neointima formation often causes structural change in the blood vessel [18], and PLD2-mediated cPA production, together with PPARγ agonists prevents neointima formation and upregulation of PPARγ target genes [2], we hypothesized that HDAC2 may be mediated by PPARγ-dependent signaling mechanisms. Furthermore, It is interesting to know if this finding is the case in human vascular smooth muscle cells (VSMC). VSMC contribute to vessel wall inflammation as well as to the formation of neointima [19]. Recent study has emphasized that cPA is effective in negatively modulating PPARγ-dependent vascular wall responses in vivo [2]. This study provide evidence that neointima formation by inhibiting VSMC proliferation is feasible.
cPA inhibited pro-inflammatory cytokine expression after AGP exposure HCAECs
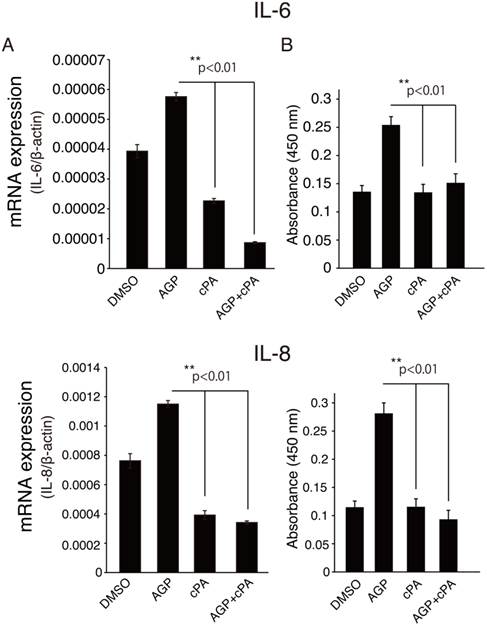
The role of AGP-induced downregulation of HDAC2 and its downstream target genes in HCAECs has not been elucidated. AGP has attracted considerable interest because of its multiple roles in physiological and pathological conditions [1]. In our previous study, we found that AGP is present in blood plasma [20] and becomes enriched after minimal oxidative modification of low-density lipoprotein (LDL). The lipid rich core in human carotid contains high concentrations of AGP, and atherothrombosis occurs in this artery upon plaque rupture [21]. As mentioned above, atherosclerosis is an inflammatory disease of the arterial wall and is therefore mediated by inflammatory cytokines. Indeed, cardiovascular disease has been shown to trigger chronic inflammatory processes, and pro-inflammatory cytokines, such as IL-6 and IL-8, are important mediators of endothelial dysfunction in coronary artery disease [22]. More recent reports have suggested that HDAC2 suppresses inflammatory gene expression [23]. Therefore, we next analyzed the expression of inflammatory mediators after treatment with or without AGP and/or cPA in HCAECs. As shown in Fig. 3A and B, AGP-treated HCAECs exhibited increased IL-6 and IL-8 mRNA expression as compared to cells treated with vehicle or cPA alone. Interestingly, when cells were cotreated with AGP plus cPA or the synthetic PPARγ antagonist T0070907, the expression of IL-6 and IL-8 mRNA was inhibited, suggesting that the effects of cPA were associated with decreased activation of PPARγ and reduced expression of IL-6 and IL-8.
AGP induced IL-6 and IL-8 expression and secretion in HCAEC culture medium. (A) Real-time PCR was used to measure the expression of IL-6 and IL-8 mRNA in HCAECs treated with vehicle or the indicated PPARγ ligand for 20 h. The relative expression of HDAC2 was calculated by normalization to β-actin expression and was expressed as the mean ± SEM (n = 3). **p < 0.01. (B) Culture supernatants were collected after 12 h of incubation with DMSO (vehicle), AGP (18:1), cPA (18:1), or AGP+cPA. Cytokine levels in the supernatants were analyzed by ELISA. Data are presented as the mean ± SEM (n = 3). **p < 0.01.
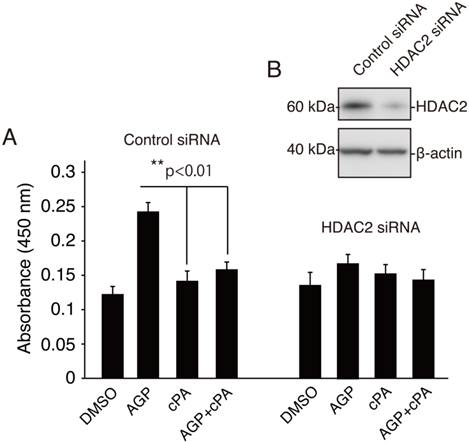
We next assessed IL-6 production by analyzing protein levels secreted into the medium from AGP- or cPA-treated HDAC2-knockdown cells. As shown in Fig. 4A and B, AGP induced increased secretion of IL-6 in cells transfected with control siRNA, and these effects were reversed by addition of cPA. However, in HDAC2-knockdown cells, AGP alone, cPA alone, or combined AGP plus cPA did not alter IL-6 secretion, suggesting that the effects of AGP on IL-6 secretion may be dependent on HDAC2 downregulation.
Knockdown of HDAC2 inhibited AGP-induced IL-6 secretion in HCAECs. (A) siRNA control-transfected HCAECs and HDAC2 siRNA-transfected HCAECs were cultured with AGP (10 μM ), cPA (10 μM ), AGP+cPA (10 μM each), or vehicle (DMSO) for 36 h. The IL-6 levels in the supernatant were analyzed by ELISA. Data are presented as the mean ± SEM (n = 3). **p < 0.01. (B) Expression of HDAC2 was knocked down in HCAECs. Total proteins were extracted from control siRNA- or HDAC2 siRNA-transfected cells. Forty-eight hours later, whole-cell lysates were subjected to western blot analysis for HDAC2. Incubation with an anti-β-actin antibody was used as protein-loading control.
Conclusion
Our findings indicated that HCAECs expressed high levels of HDAC2 and low levels HDAC1, HDAC3, and HDAC8. Moreover, AGP treatment led to reduced expression of HDAC2 and increased expression and secretion of IL-6 and IL-8 in HCAECs; these effects were reversed when cells were cotreated with cPA. Finally, siRNA-mediated silencing of HDAC2 abrogated AGP-induced IL-6 secretion, suggesting that this pathway may contribute to inflammation-mediated cardiovascular diseases. Future studies in our laboratory will focus on determining the effects of PPARγ on HDAC2 targets and their contributions to HDAC2-mediated cellular processes.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B) 25293274 (to T.T.) from the Japan Society for the Promotion of Science (JSPS), Grant-in-Aid from Takeda Science Foundation (to T.T.), and the Astellas Foundation for Research on Metabolic Disorders (to T.T.).
Abbreviations
LPA: Lysophosphatidic acid; AGP: alkyl-glycerophosphate; GPCR: G-protein-coupled receptor; PPARγ: peroxisome proliferator activated receptor gamma; HDAC: histone deacetylase; SMRT, silencing mediator of retinoid and thyroid hormone receptors; cPA: cyclic phosphatidic acid; PLD2: phospholipase D2; NCoR: nuclear receptor corepressor; NF-κB: nuclear transcription factor-κB; HCAECs: human coronary artery endothelial cells; FBS: fetal bovine serum; SDS: sodium dodecyl sulfate; TBS-T: Tris-buffered saline containing Tween 20; RT-qPCR: quantitative real-time reverse transcription polymerase chain reaction; siRNA: small-interfering RNA; DMEM: Dulbecco's modified Eagle's medium; ELISA: enzyme-linked immunosorbent assay; LDL: low-density lipoprotein.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang C, Baker DL, Yasuda S, Makarova N, Balazs L, Johnson LR. et al. Lysophosphatidic acid induces neointima formation through PPARgamma activation. J Exp Med. 2004;199:763-74
2. Tsukahara T, Tsukahara R, Fujiwara Y, Yue J, Cheng Y, Guo H. et al. Phospholipase D2-dependent inhibition of the nuclear hormone receptor PPARgamma by cyclic phosphatidic acid. Mol Cell. 2010;39:421-32
3. Tsukahara T. The Role of PPARgamma in the Transcriptional Control by Agonists and Antagonists. PPAR research. 2012;2012:362361
4. Tsukahara T, Hanazawa S, Kobayashi T, Iwamoto Y, Murakami-Murofushi K. Cyclic phosphatidic acid decreases proliferation and survival of colon cancer cells by inhibiting peroxisome proliferator-activated receptor gamma. Prostaglandins & other lipid mediators. 2010;93:126-33
5. Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril MB. et al. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Developmental cell. 2002;3:903-10
6. Fu M, Rao M, Bouras T, Wang C, Wu K, Zhang X. et al. Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. The Journal of biological chemistry. 2005;280:16934-41
7. Ye J. Improving insulin sensitivity with HDAC inhibitor. Diabetes. 2013;62:685-7
8. Gao Z, He Q, Peng B, Chiao PJ, Ye J. Regulation of nuclear translocation of HDAC3 by IkappaBalpha is required for tumor necrosis factor inhibition of peroxisome proliferator-activated receptor gamma function. The Journal of biological chemistry. 2006;281:4540-7
9. Rosenfeld MG, Glass CK. Coregulator codes of transcriptional regulation by nuclear receptors. The Journal of biological chemistry. 2001;276:36865-8
10. Gray SG, Ekstrom TJ. The human histone deacetylase family. Experimental cell research. 2001;262:75-83
11. Harris TB, Ferrucci L, Tracy RP, Corti MC, Wacholder S, Ettinger WH Jr. et al. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106:506-12
12. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135-43
13. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737-49
14. Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL. et al. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol. 2011;31:851-60
15. Kee HJ, Kook H. Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J Biomed Biotechnol. 2011;2011:928326
16. Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T. et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nature medicine. 2007;13:324-31
17. Dje N'Guessan P, Riediger F, Vardarova K, Scharf S, Eitel J, Opitz B. et al. Statins control oxidized LDL-mediated histone modifications and gene expression in cultured human endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:380-6
18. Subbotin VM. Analysis of arterial intimal hyperplasia: review and hypothesis. Theor Biol Med Model. 2007;4:41
19. Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nature medicine. 2002;8:1249-56
20. Xiao YJ, Schwartz B, Washington M, Kennedy A, Webster K, Belinson J. et al. Electrospray ionization mass spectrometry analysis of lysophospholipids in human ascitic fluids: comparison of the lysophospholipid contents in malignant vs nonmalignant ascitic fluids. Analytical biochemistry. 2001;290:302-13
21. Cheng Y, Makarova N, Tsukahara R, Guo H, Shuyu E, Farrar P. et al. Lysophosphatidic acid-induced arterial wall remodeling: requirement of PPARgamma but not LPA1 or LPA2 GPCR. Cellular signalling. 2009;21:1874-84
22. Kofler S, Nickel T, Weis M. Role of cytokines in cardiovascular diseases: a focus on endothelial responses to inflammation. Clinical science. 2005;108:205-13
23. Barnes PJ. Role of HDAC2 in the pathophysiology of COPD. Annual review of physiology. 2009;71:451-64
Author contact
![]() Corresponding author: Tamotsu Tsukahara, Department of Hematology and Immunology, Kanazawa Medical University, 1-1 Daigaku, Uchinada, Ishikawa 920-0293, Japan. ttamotsuac.jp
Corresponding author: Tamotsu Tsukahara, Department of Hematology and Immunology, Kanazawa Medical University, 1-1 Daigaku, Uchinada, Ishikawa 920-0293, Japan. ttamotsuac.jp