Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(4):356-364. doi:10.7150/ijms.7793 This issue Cite
Research Paper
Osteoclastogenic Potential of Peripheral Blood Mononuclear Cells in Cleidocranial Dysplasia
Maria Felicia Faienza1, Annamaria Ventura1, Laura Piacente1, Maria Ciccarelli1, Margherita Gigante2, Loreto Gesualdo2, Silvia Colucci3, Luciano Cavallo1, Maria Grano3, Giacomina Brunetti3 ![]()
1. Department of Biomedical Sciences and Human Oncology, Section of Pediatrics, University of Bari, Bari, Italy;
2. Nephrology, Dialysis and Transplantation Unit, Department of Emergency and Organ Transplantation, University of Bari, Bari, Italy;
3. Department of Basic Medical Sciences, Neurosciences and Sense Organs, Section of Human Anatomy and Histology, University of Bari, Bari, Italy.
Received 2013-10-1; Accepted 2014-1-10; Published 2014-2-20
Abstract
Cleidocranial dysplasia (CCD) is an autosomal dominant skeletal dysplasia characterized by hypoplastic or aplastic clavicles, dental abnormalities, and delayed closure of the cranial sutures. In addition, mid-face hypoplasia, short stature, skeletal anomalies and osteoporosis are common. We aimed to evaluate osteoclastogenesis in a child (4 years old), who presented with clinical signs of CCD and who have been diagnosed as affected by deletion of RUNX2, master gene in osteoblast differentiation, but also affecting T cell development and indirectly osteoclastogenesis. The results of this study may help to understand whether in this disease is present an alteration in the bone-resorptive cells, the osteoclasts (OCs). Unfractionated and T cell-depleted Peripheral Blood Mononuclear Cells (PBMCs) from patient were cultured in presence/absence of recombinant human M-CSF and RANKL. At the end of the culture period, OCs only developed following the addition of M-CSF and RANKL. Moreover, real-time PCR experiment showed that freshly isolated T cells expressed the osteoclastogenic cytokines (RANKL and TNFα) at very low level, as in controls. This is in accordance with results arising from flow cytometry experiments demonstrating an high percentage of circulating CD4+CD28+ and CD4+CD27+ T cells, not able to produce osteoclastogenic cytokines. Also RANKL, OPG and CTX serum levels in CCD patient are similar to controls, whereas QUS measurements showed an osteoporotic status (BTT-Z score -3.09) in the patient. In conclusions, our findings suggest that the heterozygous deletion of RUNX2 in this CCD patient did not alter the osteoclastogenic potential of PBMCs in vitro.
Keywords: Cleidocranial dysplasia, osteoclastogenesis, RUNX2
1. Introduction
Cleidocranial dysplasia (CCD, MIM#119600) is a rare skeletal disorder affecting both membranous and endochondral bone formation. Although whole skeletal system may be affected, the main clinical manifestations of CCD are malformations of the skull and clavicles which lead to the typical appearance of the face and shoulders [1]. In addition, multiple dental abnormalities, osteoporosis, delayed and deficient ossification of long bones are common [2]. The phenotypic variability observed in CCD patients is related to heterozygous alterations of Runx2 gene, a transcription factor with a prominent role in bone biology [3,4, 5]. In particular, it is essential for the commitment of pluripotent mesenchymal cells towards osteoblasts (OBs), the bone forming cells. Runx2 gene expression must be tightly regulated to support skeletal health: while sufficient activity is necessary for adequate osteogenesis, reduced or excessive Runx2 activity may lead to osteoporosis [6]. Accordingly, single nucleotide polymorphism in the human Runx2 locus has been associated with variations in bone mineral density [7]. In addition to its key role in OB differentiation, the expression of this gene has been described in T cells, as regulator of their development. In particular, in Runx2-/- embryos the development of T cells was normal in the thymus, although the number of thymocytes was significantly fewer than in wild type and heterozygous embryos [8]. Runx2 overexpression leads to an accumulation of cells at the CD8+ immature stage that were delayed in advanced differentiation [9] and predisposes to T-cell lymphomas [10].
Moreover, several in vitro studies demonstrate that Runx2 indirectly affects osteoclast (OC) formation, through the regulation of the expression of receptor activator of nuclear factor-B ligand (RANKL) and/or Osteoprotegerin (OPG) in OBs as well as in cancer and vascular smooth muscle cells [13-15]. In fact, OCs do not show significant Runx2 expression, but in Runx2-/- embryos few OCs appeared at the perichondrium, in addition their size and nuclei number are reduced in comparison with those in wild-type embryos [8]. OCs derived from the fusion of mononuclear phagocytes, the OC precursors (OCPs) [16], under the influence of two cytokines, namely macrophage-colony stimulating factor (M-CSF) and RANKL [17]. RANKL expressed on OBs and stromal cells promotes differentiation and activates mature OCs to reabsorb bone by binding to its specific receptor RANK [17]. OPG, a soluble decoy receptor secreted by OBs and bone marrow stromal cells, competes with RANK in binding to RANKL, preventing its osteoclastogenic effect [17]. RANKL and OPG are also secreted by activated T cells [18], which are critical regulators of bone turnover [19]. In particular, resting T cells have a protective role on bone as it has been shown to blunt OC formation in vitro and bone resorption in vivo [19]. In contrast, activated T cells may contribute to pathological bone loss [20-25], however distinct functions have been recognized to the different T cell subsets. In detail, CD4+ cells may sustain or inhibit osteoclastogenesis whereas, activated CD8+ cells inhibit OC formation [19, 26, 27].
Thus, because OCs can be generated from peripheral blood mononuclear cells (PBMCs) in the presence of M-CSF and RANKL, and Runx2 affect T cell development, the aim of this paper was to highlight the osteoclastogenic potential of unstimulated and unfractionated PBMCs from a 4 year old subject affected by CCD, whose we performed Runx2 genetic analysis, and to evaluate the presence of circulating OCPs as well as the role of T cell in OC formation in this skeletal disease.
2. Patient and Methods
2.1 Clinical brief
The index case was a 4-year-old male patient referred for short stature to the Endocrinology Unit of the Pediatric Clinic “B. Trambusti”, University of Bari, Italy. He was born at 35 weeks' gestation by caesarean section following an uncomplicated pregnancy. At birth: weight 2230 g (<25th percentile), length 45 cm (≤ 25th percentile), relative macrocrania (37 cm; >97th percentile) with open sutures and fontanelles (anterior fontanelle: 3·4 cm). He developed normally until 1 year of age, when his growth velocity started to decrease. Dentition was delayed (12 months of age). Psychomotor development has been normal and no apparent health problems have so far been apparent. When he presented to our observation (4 year of age) he showed a phenotype suggestive for CCD: a short and broad body build (height 91 cm, <3th percentile), a brachycephal aspect of the face with mild dysmorphisms, moderate micrognathia and a high palatal vault, persistence of large anterior fontanelle with diastasis of the sagittal suture, and sloping shoulders. A radiological survey showing a wide sagittal suture of the skull, Wormian bones near the lambdoid suture, a dysplastic right clavicle with discontinuity of the middle part with a gothic shaped thorax and hypoplastic pubic bones. Screening laboratory test for alkaline phosphatase, magnesium, free thyroxine (T4), thyroid-stimulating hormone (TSH), and calcium resulted all within normal limits. The family history identified the patient's mother with bony abnormalities, delayed ossification, and short stature (height 137 cm), who had never been undergone genetic testing for suspected CCD. A complete heterozygous gene deletion, supporting the diagnosis of CCD, was found in both mother and son. After genetic diagnosis, peripheral blood was collected from the patient to carry out cell cultures, flow cytometry analysis, real time- PCR and serum measurements of RANKL, OPG and C-terminal telopeptides of Type I collagen (CTX). The study was approved by the Ethics Committee of our Institute and informed consent was given from the patient's parents.
2.2 Bone mineral measurements
Bone quality was assessed in CCD patient and in 10 age and sex-matched healthy controls recruited from relatives of our medical staff, by quantitative ultrasound (QUS) measurements performed with a DBM Sonic 1200 bone profiler (Igea S.r.l., Carpi, MO, Italy) employing a sound frequency of 1.25 MHz. QUS is an easy, cheap and radiation-free technique to evaluate bone mineral status at peripheral skeleton through amplitude-dependent speed of sound (Ad-SoS), that expresses the ultrasound velocity inside the bone, and bone transmission time (BTT), reflecting the bone characteristics without the interference of the soft tissue. The International Society for Clinical Densitometry (ISCD) 2007 Position Development Conference (PDC) addressed clinical applications of QUS for fracture risk assessment, diagnosis of osteoporosis, treatment initiation, monitoring of treatment, and quality assurance/quality control [28].
QUS was performed on the second to the fifth proximal phalanges of the non-dominant hand and the mean value was calculated. Measurements were performed by the same operator and the coefficient of variation was 0.73%, determined by repeated measurements in the group of the 10 control subjects (three measurements per person on three different days). Ad-SoS and BTT results were expressed as Z-scores calculated on the basis of the normal values for age and sex obtained in a large Italian population sample [29].
2.3 Osteoclastogenesis
PBMCs from the CCD patients and 10 male controls (median age 4.5 ± 1.2) were isolated by gradient centrifugation with Histopaque 1077 (Sigma Aldrich, Milan, Italy), and plated at a density of 1.5 × 106 cells/cm2 in α-minimal essential medium (Gibco Limited, Uxbridge, UK), supplemented with 10% fetal bovine serum. To obtain fully differentiated human OCs, PBMCs were then cultured for about 21 days in the presence or absence of 25 ng/ml recombinant human macrophage colony stimulating factor (rh-MCSF) and 30 ng/ml rhRANKL (R&D Systems Inc, MN, USA). At the 21th day of culture, mature OCs were identified as tartrate-resistant acid phosphatase positive (TRAP +) multinucleated cells (Sigma Aldrich) containing three or more nuclei. Moreover, their resorbing activity was demonstrated by plating the cells on multiwell slides (4 × 105 cells/well) coated with a calcium phosphate film (Millenium Osteologic; Millenium Biologix Inc, Ontario, Canada). This system incorporates a resorbable artificial bone in the form of submicron calcium phosphate films. To visualize the resorption areas formed by the OCs, after 21 days of culture, the cells were removed by adding NaOCl to each well. The slides were washed with double distilled water (pH 7) three times. This procedure was repeated three to five times until the slides were completed cleaned of cell fragments, then the percentage of mineral surface resorbed by the OCs was quantified with an image analyser. The photomicrographs of OCs in cultures were obtained using a Nikon Eclipse E400 microscope equipped with a Nikon plan fluor 10 × /030 DICL. The microscope was connected to a Nikon digital camera (DXM 1200); the acquisition was performed using the software LUCIA G version 4.61 (BUILD 0.64) for Nikon, Italy. T cell-depletion was performed using anti-CD2 antibody coated immunomagnetic Dynabeads (Dynal, Lake Success, NY, USA). T cell-depleted PBMCs were cultured with or without exogenous cytokines. The degree of T cell-depletion was checked by flow cytometry analysis.
2.4 Flow cytometry analysis
Freshly EDTA peripheral blood from controls and patients were stained with suitable conjugated antibody: APC-CD8, FITC-CD4, PE-CD28, Pe-Cy-5-CD27, Pe-Cy-5-CD25, PE-CD62L, PE-CD69, Pe-Cy-5-CD14 and PE-CD16 (all Beckmann Coulter, Milan, Italy). Cells were incubated for 15 min at room temperature and for other 10 minute with VersaLyse reagent (Beckmann Coulter) to lyse red blood cells. Data were acquired using a FC500 (Beckmann Coulter) flow cytometer and analyzed using Kaluza software. The area of positivity was determined using an isotype-matched mAb, a total of 104 events for each sample were acquired.
2.5 RNA isolation and real-time PCR
Total RNA was extracted from CD2+ and CD14+ cells isolated from controls and CCD patient using spin columns (RNeasy, QIAGEN, Hilden, Germany), according to the manufacturer's instructions. The extracted RNA was reverse-transcribed using the Super Script First-Stand Synthesis System kit for RT-PCR (Life Technologies, Carlsbard, CA, USA): an RT mixture containing 1µg total RNA, dNTPs (deoxyribonucleoside triphosphates), Oligo(dT), RT buffer, MgCl2, DTT (dithiothreitol), RNaseOUT, SuperScript II RT, DEPC (diethyl pyrocarbonate)-treated water to final volume 100µL was prepared, according to the manufacturer's instructions. cDNA was amplified with the iTaq SYBR Green supermix with ROX kit (Bio-Rad Laboratories, Bio-Rad Laboratories Inc., Hercules, CA, USA) and the PCR amplification was performed using the Chromo4 Real-Time PCR Detection System (Bio-Rad Laboratories). The following primer pairs were used: RANKL S: CGTTGGATCACAGCACAT, RANKL AS: GCTCCTCTTGGCCAGTC; TNF-alpha S: ATCTACTCCCAGGTCCTC, TNF-alpha AS: GATGCGGCTGATGGTGT; RANK S: CAGGATGCTCTCATTGGTCAG, RANK AS: AGAAAGGAGGTGTGGATTGC; c-fms S: GTGGCAGGAAGGTGATGTC, c-fms AS: GCTTGGTGTTGTTGTGTTGG; Ciclophylin S: 5'-CAGGTCCTGGCATCTTGTCC-3'; Ciclophylin AS: 5'-TTGCTGGTCTTGCCATTCCT-3'.
The running conditions were as follows: incubation at 95°C for 3 min, and 40 cycles of incubation at 95°C for 15 s and 60°C for 30 s. After the last cycle, the melting curve analysis was performed into 55-95°C intervals by incrementing the temperature of 0.5°C. The fold change values were calculated by Pfaffl's method [30].
2.6 ELISA
sRANKL, OPG (Biomedica, Vienna, Austria), bone-specific alkaline phosphatase (Dade Behring Inc, Newark, DE) and C-terminal telopeptides of Type I collagen (CTX) (Serum CrossLaps, Immunodiagnostics Systems Inc, Fountain Hills, AZ, USA) were measured in the sera obtained from CCD patient as well as in the sera of aged-matched healthy donors using commercially available ELISA kit, according to the manufacturer's instructions. The absorption was determined with an ELISA reader, 550 Microplate Reader (Bio-Rad), and the results were expressed as mean ± SE. Parathyroid hormone (PTH) (IMULITE, American System), bone alkaline phosphatase (Dade Behring Inc, Newark, DE) and osteocalcin (IBL International GMBH, Hamburg, Germany) were also measured.
2.7 Statistical analyses
Statistical analyses were performed by student T test with the Statistical Package for the Social Sciences (spssx/pc) software (SPSS, Chicago, IL, USA). The results were considered statistically significant for p<0.05.
3. Results
3.1 Osteoclastogenesis in CCD patient
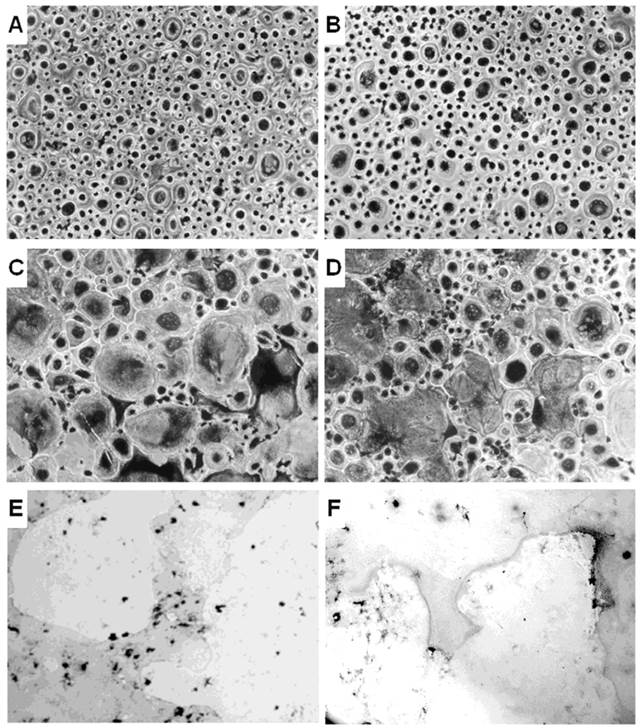
The unfractionated PBMCs from CCD patient and controls were cultured without or with the osteoclastogenic cytokines MCSF and RANKL. Few small TRAP+ OCs were identified in the unstimulated PBMC cultures from controls and CCD patient (Figure 1A, 1B). Although the OC number is higher in patient than in the controls, the difference did not reach a statistical significance (OC average number/well = 15 ± 5 and 10 ± 3, respectively, p<0.19). Moreover, the addition of MCSF and RANKL to PBMC cultures from control and CCD patient significantly increased the OC number (OC average number/well = 73 ± 4 and 69 ± 6, respectively; P < 0.02; Figure 1C, 1D), compared with the parallel unstimulated cultures. No statistical significant differences in the OC number was measured in PBMC culture treated with MCSF and RANKL between the two groups. Moreover, the OCs formed in the presence of the osteoclastogenic cytokines are active in bone resorption for both controls and CCD patient (Figure 1E, 1F).
Osteoclastogenesis in CCD patient. OCs were obtained from unfractionated PBMCs of controls (A, C, E) and CCD patient (B, D, F). Rare and small-sized OCs were observed in the unstimulated cultures from controls (A) and patient (B). Exogenous M-CSF and RANKL were essential to triggering the OC formation in controls (C) and patients (D). Multinucleated (> 3 nuclei per cell) and TRAP+ cells were identified as OCs (magnification, × 200). Photomicrographs of the pits formed on Millennium Osteologic slides by the OCs (E, F). Large resorption areas were observed in both control (E) and patient (F) cultures treated with M-CSF and RANKL.
3.2 Monocytes and T cells in CCD patient
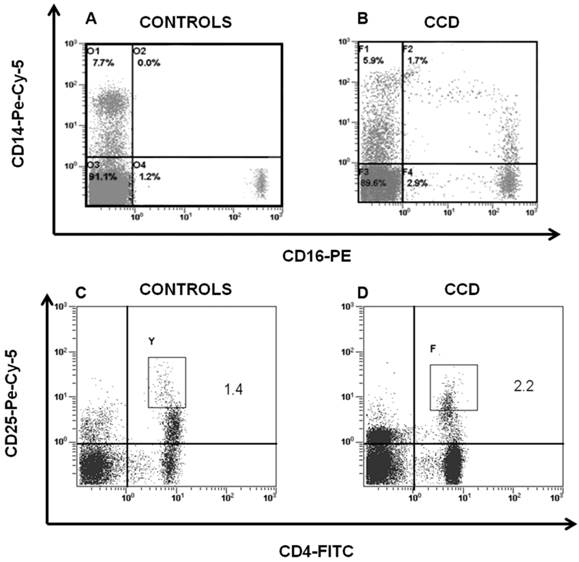
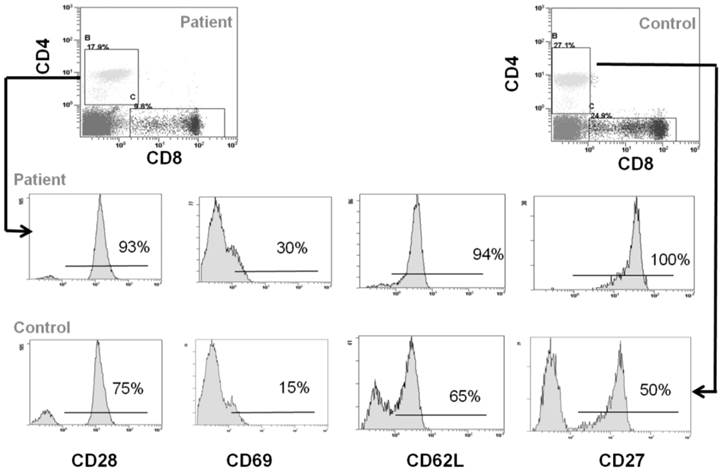
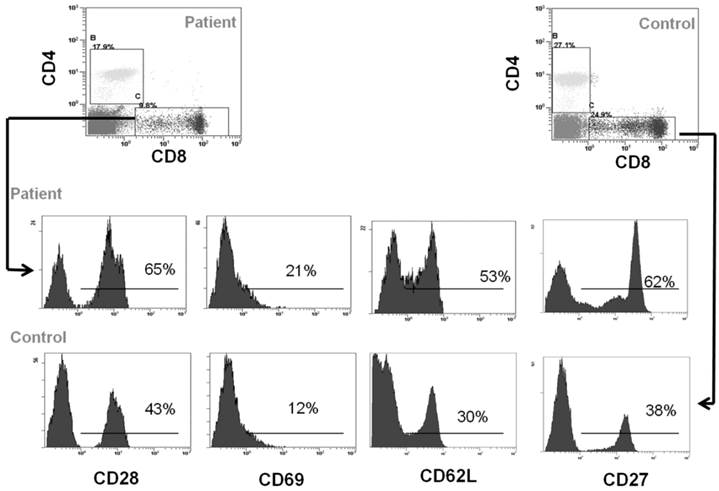
Peripheral blood cells from CCD patient and controls were subjected to flow cytometry analysis to evaluate the immunophenotype of monocytes and T cells. In particular, in the patient we found a percentage (1.7 ± 0.5%) of CD14+/CD16+ cells, which can be considered committed OC precursors [31], that are absent in controls (P < 0.01), Figure 2A-2B. Moreover, we characterized both CD4+ and CD8+ T cells focusing on markers of T-cell activation, proliferation, and differentiation. No significant differences for CD4+CD25high population were observed between patient and controls (Figure 2C-2D). Staining of freshly peripheral blood T cells revealed that costimulatory molecules (CD28, CD27, CD62L, CD69) were highly expressed on both CD4+ and CD8+ T cells cells from CCD patient (Figure 3-4). Moreover, we also showed that in CCD patient the subset CD4+/CD28- is smaller than controls (Figure 3). This is an important finding because this subset is involved in the production of TNFα, and consequently supporting osteoclastogenesis. Similarly, we also found that the subset CD8+/CD27- include a small percentage of cells in patient (Figure 4). This is relevant because this subset is involved in the production of inflammatory cytokines.
CD14+/CD16+ and CD4+/CD25high cells in CCD patient. Flow cytometry analysis of CD14+/CD16+ and CD4+/CD25high cells in peripheral blood cells revealed that CD14+/CD16+ was significantly higher in patient (B) compared with the age-matched normal controls (A), whereas no significant differences were detected in CD4+/CD25high cells among patient (D) and controls (C).
Immunophenotype of CD4+ T cells. Flow cytometry analysis of CD28, CD69, CD62L and CD27 in CD4+ cells from CCD patient and controls.
Immunophenotype of CD8+ T cells. Flow cytometry analysis of CD28, CD69, CD62L and CD27 in CD4+ cells from CCD patient and controls.
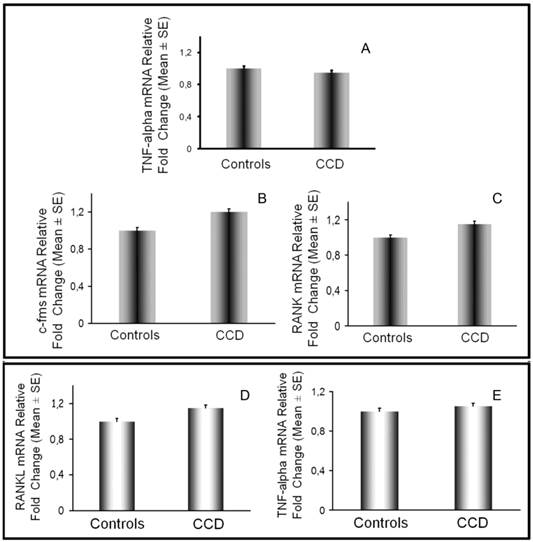
On monocytes we also evaluated at mRNA level the expression of c-fms and RANK, respectively receptor for MCSF and RANK, and we did not find differences between patient and controls (Figure 5B-5C). According to these results, cultures of T cell-depleted PBMCs from CCD patient required MCSF and RANKL to lead OC formation, as in controls (data not shown). Additionally, we analyzed the expression of the osteoclastogenic factors by T cells and monocytes. In CCD patient and controls, fresh T cells purified from PBMCs, expressed similar RANKL and TNFα levels as demonstrated by real time PCR (Fig. 5D-5E) and flow cytometry (data not shown). Monocytes from CCD patient, also expressed TNFα at the same levels respect to the controls (Fig. 5A).
Monocyte and T cell characterization by real-time PCR. TNFα, c-fms and RANK expression was evaluated in monocytes (A, B, C) by real time PCR. RANKL (D) and TNFα (E) mRNA levels were assessed in T cells from patient and control No significant differences were detected in their expression among CCD patient and controls.
3.3 Bone and Serum measurements
As for the results of QUS, mean Ad-SoS Z-score as well as mean BTT Z-score were normal in healthy controls (0.01 ± 1.0 and -0.03 ± 0.8, respectively). In CCD patient AdSos Z-score was -1.56, and BTT Z-score was -3.09, thus revealing osteoporosis. The quantification of CTX (0.691 ± 0.005 vs 0.411 ± 0.09 ng/mL, range 0.151-0.870 ng/mL), indicator of bone resorption, as well as of PTH (20 ± 0.020 pg/ml vs 23 ± 5.62) indicated that patient had similar levels to the control group. We found that sRANKL levels are undetectable, as in many controls; however lower respect the complete control group (0.05 ± 0.02 pmol/L). Otherwise, in CCD patient OPG level is comprised in the range of the control group (0.45 ± 0.02 pmol/L vs 0.50 ± 0.22 pmol/L). In CCD patient, serum levels of osteocalcin and bone alkaline phosphatase, markers of OB activity, were in the normal range, 18.5 ± 0.03 U/L and 139 ± 5.6 U/L, respectively.
4. Discussion
The current study demonstrated that in a young CCD patient, with serious osteoporosis, numerous skeletal abnormalities and complete heterozygous deletion of Runx2 gene osteoclastogenesis in vitro occurred as in controls. These results suggest that the deletion of Runx2 directly does not impair OCPs, OC formation and activity in vitro. However, we cannot completely exclude that osteoclastogenesis might be indirectly affected by this deletion in vivo, as OBs supported this process, although we found that serum levels of CTX, bone resorption marker, are in the control range. On the other hand, although the patient has the complete heterozygous deletion of Runx2, master gene of OB differentiation, we found that serum markers of OB activity as bone alkaline phosphatase and osteocalcin are in the control range. According to our results, other authors demonstrated that alkaline phosphatase function is disturbed in only serious cases, in which CCD is also associated to hypophosphatasia [32, 33]. The major limitation of our study is that the results refer to a single patient, seeing as the CCD is a rare skeletal dysplasia.
In literature, there are few data about bone cells activity in CCD patients; however interesting findings are available on in vivo and in vitro models. In detail, the OC formation and activity was evaluated in heterozygous Runx2 mice that mimicked most of the abnormalities of CCD patients [34]. In fact, they have delayed tooth eruption that appear to be linked to the fact that heterozygous mice cannot recruit sufficient OCs for the alveolar bone resorption, which is essential for tooth eruption. In particular, the authors showed that the values of Oc.S/BS in these mice were lower than that of wild-type mice only in the eruption area and not in femora [35]. Gao YH et al., using a murine culture system of calvarial and spleen cells treated with 1α,25(OH)2vitaminD3 and dexametasone, reported that co-culture of Runx2-/- calvarial cells generated a significantly lower number of osteoclastic cells in comparison with those generated in co-cultures with wild-type calvarial cells [36]. Accordingly, in Runx2-/- embryos few and small OCs appeared at the perichondrium in comparison with those in wild-type embryos [8]. In periodontal ligament (PDL) cells from CCD patients there was a reduced basal expression of mRNA for the key regulatory gene for bone remodeling RANKL [37]. Furthermore, compared to the control cells from healthy donors, it was less inducible by stimulation of the cultures with 1α, 25(OH)2D3 [37]. Moreover, compared to the control group of healthy donors, the PDL cells of the patients with CCD showed a comparable mRNA expression of OPG, as well as an inhibitory effect of 1,25(OH)2D3. Consequently, the ratio of RANKL to OPG was decreased in the cultures of the patients with CCD, thus leading to the inhibition of bone resorption [37]. In fact, the conditioned medium of cultures of the PDL cells of patients with CCD lead to reduced differentiation of TRAP-positive multinucleated cells and the data of co-culture experiments showed inhibited expression of typical OC markers when PDL cells of patients with CCD were co-cultivated with RAW cells and stimulated with 1,25(OH)2D3 [37]. These findings were in line with our findings showing undetectable sRANKL levels in patient serum respect to OPG, thus the RANKL/OPG ratio shifted in favor of OPG, even if this condition is also typical of controls.
Interestingly, in our CCD patient although monocytes express c-fms and RANK at the same level of controls, CD14+/16+ monocytes, OCPs with a prominent role in erosive arthritis [31], are increased respect to the controls, however, in vitro this condition is not sufficient to drive OC formation in absence of osteoclastogenic cytokines. In fact, spontaneously they did not differentiate in OCs because T cells did not produce cytokines, such as RANKL or TNFα, sustaining the process. These findings are supported by the evaluation of immunophenotype of T cells, which highlight an activation status of T cells, but not of subsets involved in osteoclastogenesis support. In detail, in CCD patient the subset CD4+/CD28- is smaller than in controls. This is an important finding because this subset is involved in the production of TNFα [38]. Similarly, the subset CD8+/CD27- include a small percentage of cells in patient [39]. This is significant because this subset might be involved in the production of inflammatory cytokines, with a prominent role in sustaining osteoclastogenesis. However, activated CD8+ cells inhibit OC formation or activity [26, 27], according to our findings. It seems that the heterozygous deletion of Runx2 has a protective role towards osteoclastogenesis driven by T cells in the patient. Consistent with these observations, it is known that there is a site for Runx2 binding in the regulatory region of many T cell specific genes with consequent Runx2-linked T cell-specific transcriptional regulation.
5. Conclusion
The present study showed for the first time that in a young CCD patient with serious osteoporosis, numerous skeletal abnormalities and complete Runx2 heterozygous deletion osteoclastogenesis in vitro occurred as in controls, suggesting that this deletion directly does not alter the osteoclastogenic potential of PBMCs in vitro. However, we cannot exclude that in vivo the impairment of OBs from CCD patient, might indirectly also alter OC activity.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gorlin RJ, Cohen MM, Levin LS. Syndromes affecting bone: other skeletal disorders, in: Syndromes of the head and the neck; 3rd edn. Oxford: Oxford University Press. 1990:249-253
2. Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet. 1999;36(3):177-182
3. Otto F, Kanegane H, Mundlos S. Mutations in the RUNX2 gene in patients with cleidocranial dysplasia. Hum Mutat. 2002;19(3):209-216
4. Zhou G, Chen Y, Zhou L, Thirunavukkarasu K, Hecht J, Chitayat D, Gelb BD, Pirinen S, Berry SA, Greenberg CR, Karsenty G, Lee B. CBFA1 mutation analysis and functional correlation with phenotypic variability in cleidocranial dysplasia. Hum Mol Genet. 1999;8(12):2311-2316
5. Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15(4):467-481
6. Liu W, Toyosawa S, Furuichi T, Kanatani N, Yoshida C, Liu Y, Himeno M, Narai S, Yamaguchi A, Komori T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J Cell Biol. 2001;155(1):157-166
7. Vaughan T, Pasco JA, Kotowicz MA, Nicholson GC, Morrison NA. Alleles of RUNX2/CBFA1 gene are associated with differences in bone mineral density and risk of fracture. J Bone Miner Res. 2002;17(8):1527-1534
8. Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755-764
9. Vaillant F, Blyth K, Andrew L, Neil JC, Cameron ER. Enforced expression of Runx2 perturbs T cell development at a stage coincident with beta-selection. J Immunol. 2002;169(6):2866-2874
10. Vaillant F, Blyth K, Terry A, Bell M, Cameron ER, Neil J, Stewart M. A full-length Cbfa1 gene product perturbs T-cell development and promotes lymphomagenesis in synergy with myc. Oncogene. 1999;18(50):7124-7134
11. Satake M, Nomura S, Yamaguchi-Iwai Y, Takahama Y, Hashimoto Y, Niki M, Kitamura Y, Ito Y. Expression of the Runt domain-encoding PEBP2 alpha genes in T cells during thymic development. Mol Cell Biol. 1995;15(3):1662-1670
12. Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila runt gene and the human AML1 gene. Proc Natl Acad Sci USA. 1993;90(14):6859-6863
13. Jonason JH, Xiao G, Zhang M, Xing L, Chen D. Post-translational Regulation of Runx2 in Bone and Cartilage. J Dent Res. 2009;88(8):693-703
14. Gupta A, Cao W, Chellaiah MA. Integrin αvβ3 and CD44 pathways in metastatic prostate cancer cells support osteoclastogenesis via a Runx2/Smad 5/receptor activator of NF-κB ligand signaling axis. Mol Cancer. 2012;11:66
15. Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, Anderson PG, Tintut Y, Demer LL, Wang D, Chen Y. Runx2-upregulated receptor activator of nuclear factor κB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. 2011;31(6):1387-1396
16. Massey HM, Flanagan AM. Human osteoclasts derive from CD14-positive monocytes. Br J Haematol. 1999;106(1):167-170
17. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337-342
18. Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002;20:795-823
19. Mori G, D'Amelio P, Faccio R, Brunetti G. The interplay between the bone and the immune cells. Clin Dev Immunol. 2013:ArticleID720504
20. Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402(6759):304-309
21. Colucci S, Brunetti G, Rizzi R, Zonno A, Mori G, Colaianni G, Del Prete D, Faccio R, Liso A, Capalbo S, Liso V, Zallone A, Grano M. T cells support osteoclastogenesis in an in vitro model derived from human multiple myeloma bone disease: the role of the OPG/TRAIL interaction. Blood. 2004;104(12):3722-3730
22. Faienza MF, Brunetti G, Colucci S, Piacente L, Ciccarelli M, Giordani L, Del Vecchio GC, D'Amore M, Albanese L, Cavallo L, Grano M. Osteoclastogenesis in children with 21-hydroxylase deficiency on long-term glucocorticoid therapy: the role of receptor activator of nuclear factor-kappaB ligand/osteoprotegerin imbalance. J Clin Endocrinol Metab. 2009;94(7):2269-2276
23. Brunetti G, Colucci S, Pignataro P, Coricciati M, Mori G, Cirulli N, Zallone A, Grassi FR, Grano M. T cells support the osteoclastogenesis in an in vitro model derived from human periodontitis patients. J Periodontol. 2005;76(10):1675-1680
24. Colucci S, Brunetti G, Cantatore FP, Oranger A, Mori G, Quarta L, Cirulli N, Mancini L, Corrado A, Grassi FR, Grano M. Lymphocytes and synovial fluid fibroblasts support the osteoclastogenesis through RANKL, TNFα, and IL-7 in an in vitro model derived from human psoriatic arthritis. J Pathol. 2007;212(1):47-55
25. Colucci S, Brunetti G, Mori G, Oranger A, Centonze M, Mori C, Cantatore FP, Tamma R, Rizzi R, Liso V, Zallone A, Grano M. Soluble decoy receptor 3 (DcR3) modulates the survival and formation of osteoclasts from multiple myeloma bone disease patients. Leukemia. 2009;23(11):2139-2146
26. Choi Y, Woo KM, Ko SH, Lee YJ, Park SJ, Kim HM, Kwon BS. Osteoclastogenesis is enhanced by activated B cells but suppressed by activated CD8(+) T cells. Eur J Immunol. 2001;31(7):2179-88
27. Buchwald ZS, Kiesel JR, DiPaolo R, Pagadala MS, Aurora R. Osteoclast activated FoxP3+ CD8+ T-cells suppress bone resorption in vitro. PLoS One. 2012;7(6):e38199
28. Krieg MA, Barkmann R, Gonnelli S, Stewart A, Bauer DC, Del Rio Barquero L, Kaufman JJ, Lorenc R, Miller PD, Olszynski WP, Poiana C, Schott AM, Lewiecki EM, Hans D. Quantitative ultrasound in the management of osteoporosis: the 2007 ISCD Official Positions. J Clin Densitom. 2008;11(1):163-187
29. Baroncelli GI, Federico G, Vignolo M, Valerio G, del Puente A, Maghnie M, Baserga M, Farello G, Saggese G; Phalangeal Quantitative Ultrasound Group. Cross-sectional reference data for phalangeal quantitative ultrasound from early childhood to young adulthood according to gender, age, skeletal growth, and pubertal development. Bone. 2006;39(1):159-173
30. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45
31. Chiu YG, Shao T, Feng C, Mensah KA, Thullen M, Schwarz EM, Ritchlin CT. CD16 (FcRgammaIII) as a potential marker of osteoclast precursors in psoriatic arthritis. Arthritis Res Ther. 2010;12(1):R14
32. Morava E, Kárteszi J, Weisenbach J, Caliebe A, Mundlos S, Méhes K. Cleidocranial dysplasia with decreased bone density and biochemical findings of hypophosphatasia. Eur J Pediatr. 2002;161(11):619-622
33. Unger S, Mornet E, Mundlos S, Blaser S, Cole DE. Severe cleidocranial dysplasia can mimic hypophosphatasia. Eur J Pediatr. 2002;161(11):623-626
34. Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765-771
35. Yoda S, Suda N, Kitahara Y, Komori T, Ohyama K. Delayed tooth eruption and suppressed osteoclast number in the eruption pathway of heterozygous Runx2/Cbfa1 knockout mice. Arch Oral Biol. 2004;49(6):435-442
36. Gao YH, Shinki T, Yuasa T, Kataoka-Enomoto H, Komori T, Suda T, Yamaguchi A. Potential role of cbfa1, an essential transcriptional factor for osteoblast differentiation, in osteoclastogenesis: regulation of mRNA expression of osteoclast differentiation factor (ODF). Biochem Biophys Res Commun. 1998;252(3):697-702
37. Lossdörfer S, Abou Jamra B, Rath-Deschner B, Götz W, Abou Jamra R, Braumann B, Jäger A. The role of periodontal ligament cells in delayed tooth eruption in patients with cleidocranial dysostosis. J Orofac Orthop. 2009;70(6):495-510
38. Bryl E, Vallejo AN, Weyand CM, Goronzy JJ. Down-regulation of CD28 expression by TNF-α. J. Immunol. 2001;167(6):3231-3238
39. Kohler S, Thiel A, Rudwaleit M, Sieper J, Braun J. CD27+ memory and CD27- effector CD8+ T cells are responsible for a decreased production of proinflammatory cytokines in HLA B27-positive subjects. Clin Exp Rheumatol. 2005;23(6):840-846
Author contact
![]() Corresponding author: Brunetti Giacomina, PhD. Department of Basic Medical Sciences, Neurosciences and Sense Organs, Section of Human Anatomy and Histology, University of Bari, Piazza Giulio Cesare, 11, 70124 Bari, Italy. Tel +39 0805478306 Fax: +39 0805478361 giacomina.brunettiit.
Corresponding author: Brunetti Giacomina, PhD. Department of Basic Medical Sciences, Neurosciences and Sense Organs, Section of Human Anatomy and Histology, University of Bari, Piazza Giulio Cesare, 11, 70124 Bari, Italy. Tel +39 0805478306 Fax: +39 0805478361 giacomina.brunettiit.