Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(13):1860-1867. doi:10.7150/ijms.6460 This issue Cite
Research Paper
Identification of Hypermethylation in Hepatocyte Cell Adhesion Molecule Gene Promoter Region in Bladder Carcinoma
Jia Tao1#, Qi Liu1#, Xiaohou Wu2, Xin Xu1, Yanyi Zhang1, Qiuju Wang1, Chunli Luo1 ![]()
1. Department of Laboratory Diagnosis, Chongqing Medical University, Chongqing, 400016 China;
2. Department of Urinary Surgery, First Hospital of Chongqing Medical University, Chongqing, 400016 China.
# The authors contributed equally to this work and share first authorship.
Received 2013-4-12; Accepted 2013-6-18; Published 2013-11-11
Abstract
Background: Epigenetic regulation such as aberrant hypermethylation of CpG islands in promoter plays a key role in tumorigenesis. 5-Aza-2'-deoxycytidine (5-aza-CdR) which is a potent inhibitor of DNA methylation can reverse the abnormal hypermethylation of the silenced tumor suppressor genes (TSGs). It has been reported that hepatocyte cell adhesion molecule (hepaCAM) acts as a tumor suppressor gene and expression of its mRNA and protein were down-regulated in bladder cancer. Over-expression of hepaCAM can inhibit cancer growth and arrest renal cancer cells at G0/G1 phase. In this study, we investigated the methylation status of hepaCAM gene, as well as the influence of 5-aza-CdR on expression of hepaCAM gene in bladder cancer cells. Methods: CpG islands in hepaCAM promoter and methprimers were predicted and designed using bioinformatics program. Methylation status of hepaCAM promoter was evaluated in bladder cancer tissues and two cell lines (T24 and BIU-87) by Methylation-specific PCR; Western blot and Immunofluorescence were used to detect expression of hepaCAM protein after 5-aza-CdR treatment; Flow cytometry assay was performed to determine effectiveness of 5-aza-CdR on cell cycle profile. Results: CpG island in promoter of hepaCAM gene was hyper-methylated both in bladder carcinoma tissues and cell lines (T24 and BIU-87). Otherwise, aberrant methylation of its promoter was associated with its decreased expression. Hypermethylation of hepaCAM gene was reversed and expression of its mRNA and protein were re-activated in two cell lines by DNA methyltransferases inhibitor 5-aza-CdR. Flow cytometry assay demonstrated that 5-aza-CdR can inhibit growth of cancer cells by arresting cancer cells at G0/G1 phase. Conclusion: Abnormal hypermethylation in CpG island of hepaCAM promoter is involved in absence of hepaCAM gene expression when bladder cancer occurs. Re-activation of hepaCAM gene by 5-aza-CdR can inhibit growth of cancer cells and arrest cells at G0/G1 phase.
Keywords: hepaCAM, promoter, methylation, 5-aza-CdR, Bladder carcinoma.
Introduction
Hepatocyte cell adhesion molecule (hepaCAM) was discovered by Shali Shen in 2005[1]. It is a new type of cell adhesion molecules, belonging to the immunoglobulin superfamily. Earlier researchers found that the expression of hepaCAM in bladder cancer cells and tissues was decreased or absent, and over-expression of hepaCAM can inhibit tumor growth[2,3]. For its anti-cancer ability, hepaCAM has been presumed as a tumor suppressor gene. However, the mechanism of hepaCAM gene silencing in cancer is unknown.
DNA methylation is a covalent and reversible chemical modification, resulting from the addition of a methyl (CH3) group at the carbon 5 position of the cytosine ring[4]. Aberrant DNA methylation plays an important role in regulating expression and biological function of TSGs(tumor suppressor genes), and contributing to tumorigenesis. Abundant previous studies showed that abnormal hypermethylation of CpG islands in TSGs promoter were devoted to genes silencing[5], such as RASSF1A, DAPK1, TFPI-2, p16 and CDH1[6-9]. 5-aza-CdR (5-Aza-2'-deoxycytidine) which is a DNA methyltransferases inhibitor has been proved to re-activate the expression of TSGs. For example, 5-aza-CdR induced re-expression of E-cadherin, p15 and p73 gene in cancers[10,11]. For its potent anticancer ability, 5-aza-CdR has been used for treatment of certain leukemia[12].
In this study, we have detected aberrant hypermethylation in promoter CpG islands of hepaCAM gene for the first time. Our results also indicated that hypermethylation of hepaCAM promoter significantly associated with decreased expression of its protein in bladder cancer. DNA methyltransferases inhibitor 5-aza-CdR can reverse its hypermethylation and lead to re-expression of hepaCAM gene.
Materials and Methods
Cell lines and Patients
T24 cell line was kindly donated by infectious diseases institute of Chongqing Medical University, China. BIU-87 cell line was purchased from cell bank of Wuhan University. They were cultured in RPMI1640 (Gibco, American) with 10% fetal bovine serum (Gibco, American) at 37℃ in a humidified atmosphere containing 5% CO2 incubator. 30 cases of paired urothelial carcinoma samples and corresponding adjacent tissues were collected from the department of Urology at the First Affiliated Hospital of Chongqing Medical University, with patients' consent and institutional ethics committee. The tumor grades were evaluated according to the WHO standards (I: 5, II: 22, III: 3) and staging according to the TNM classification guidelines (Ta ~ T1, 18; T2 ~ T4, 12).
Adenovirus Infection
Cells were infected by advenovirus vetor and divided into the blank control group, the negative control group (infected with empty vetor pAdH5) and the positive group (infected with pAdH5-hepaCAM).
Promoter CpG islands prediction of hepaCAM
Promoter prediction of hepaCAM gene was performed by FirstEF software[13] (http://rulai.cshl.org/tools/FirstEF/). Then, CpG islands was predicted and methprimers for MSP was designed through the bioinformatics program[14] (http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi).
Methylation-specific PCR
Genomic DNA from cells and tissues was extracted according to the TIANamp Genomic DNA Kit's instruction (TIANGEN BIOTECH,China). The bisulfite modification procedure was carried out using EZ DNA Methylation-Gold™ Kit (Zymo Research). 500ng of genomic DNA was used in bisulfite treatment and the bisulfite-modified DNA was stored at -80℃. Amplification was started in a 20ul reaction volume including 10ul Hot start PCR mix (BIOTAKE, China), 1ul of each primer, 1ul of modified DNA and 7ul of ddH2O. The cycling conditions were performed by initial denaturation at 95℃ for 5min, followed by 40 cycles of 95℃ for 50s, 58.4℃(M)/61.8℃(U) for 50s, 72℃for 1min, and a final extension at 72℃ for 10min. Primers for methylated DNA of hepaCAM were: Left M primer, 5'-AGAATTCGGTTTCGGAGTTTC-3'; Right M primer, 5'-CTAAACGACGACGAATATATCCG-3'. Primers for unmethylated DNA were: Left U primer, 5'-AGAATTTGGTTTTGGAGTTTTGA-3'; Right U primer, 5'- ACCCTAAACAACAACAAATATATCCA-3'. The product sizes of the methylated and unmethylated amplification were 172bp and 175bp, respectively. The products were visualized in a 3% polyacrylamide gel.
Treatment with 5-aza-CdR
5-aza-CdR was purchased from Sigma (USA). It dissolved in dimethylsulphoxide (DMSO) at a stock concentration of 3mmol/L and stored at -80℃. The selection of drug concentrations was referring to previous MTT result[15]. Various concentrations of 5-Aza-CdR (Table 1) were used for 72h in the treatment. DMSO was used as a control.
The experiment grouping.
| T24 | BIU-87 | |
|---|---|---|
| 1 | DMSO | DMSO |
| 2 | Blank | Blank |
| 3 | 0.3μmol/L 5-aza-CdR | 0.1μmol/L 5-aza-CdR |
| 4 | 1μmol/L 5-aza-CdR | 0.5μmol/L 5-aza-CdR |
| 5 | 3μmol/L 5-aza-CdR | 1μmol/L 5-aza-CdR |
| 6 | 10μmol/L 5-aza-CdR | 5μmol/L 5-aza-CdR |
Western-blot for hepaCAM assay
Total protein was extracted from bladder cancer cell and tissue using lysis buffer with 1mM Phenylmethanesulfonyl fluoride (PMSF). It was denatured in sodium dodecylbenzene sul fonate (SDS) loading buffer at 100℃ for 5min. Protein was electrophoresed in 12% SDS-polyacrylamide gel and transfered to polyvinylidene membranes (PVDF) (Milliproe). The membranes were blocked with 5% skim milk in TBST and then were immunoblotted overnight at 4℃ using anti-hepaCAM antibody (Proteintech, American), β-actin antibody (Santa Cruz,CA). The membranes were incubated with a peroxidase-conjugated goat anti-rabbit or mouse sencondary antibody for 1h at room temperature after being washed by TBST. Proteins were detected using a chemiluminescence (ECL) reagent (Milliproe) by western blotting detection and analysis system.
Immunofluorescence analysis
Cells cultured on coverslips were treated with 5-aza-CdR, and then fixed with 4% paraformaldehyde for 10minutes. They were blocked in 5% goat serum for 1-2h at room temperature before permeabilized with 0.1% Triton X-100 for 15 minutes. Anti-hepaCAM antibody was incubated overnight at 4℃ followed by incubation of FITC-labeled secondary antibody for 1h at room temperature in a moist container in dark. After washing with Phosphate Buffered Saline (PBS), slides were nuclear stained with 4,6-diamino-2-phenyl indole (DAPI). Fluorescence was visualized by fluorescence microscopy.
Flow cytometry assay
After treatment with 5-aza-CdR and infection with adenovirus, cells were harvested and washed with PBS three times. Cells fixed with 70% ethanol were centrifuged at 1000×g for 10min to remove ethanol, resuspended in staining solution (1×PBS, 0.1%Triton X-100,20ug/ml propidium iodide and 100ug/ml RNase A) and incubated for 30min at room temperature. Cells were subjected to cell cycle analysis by FACS.
Statistical analysis
All the statistical analysis was performed with SPSS 16.0 software using One-way ANOVA, Paired-Samples T test and Spearman correlation analysis. P<0.05 was considered statistically significant.
Results
Effect of ectogenic hepaCAM gene on cell cycle profile
Cell cycle profiles were analyzed by FCM (Fig.1). Overexpression of ectogenic hepaCAM in positive infected group arrested cells in G0/G1-phase (p<0.05). There were no statistically significant difference between the pAdH5 cells and blank cells (p>0.05) (Table 2).
Effect of ectogenic hepaCAM gene on cell cycle distribution. FCM measured the influence of pAdH5-hepaCAM on cell cycle distribution in two cell lines (T24 and BIU-87). Data are reported as mean±SD (n=3). Cell percentage in G0/G1 phase was increased in pAdH5-hepaCAM group versus blank and pAdH5 group. The results revealed that hepaCAM can arrest cell cycle at G0/G1. #: p<0.05, ##: p<0.01. (Blank: cells without treatment; pAdH5: Adenovirus empty vector; pAdH5-hepaCAM: hepaCAM adenovirus vector).
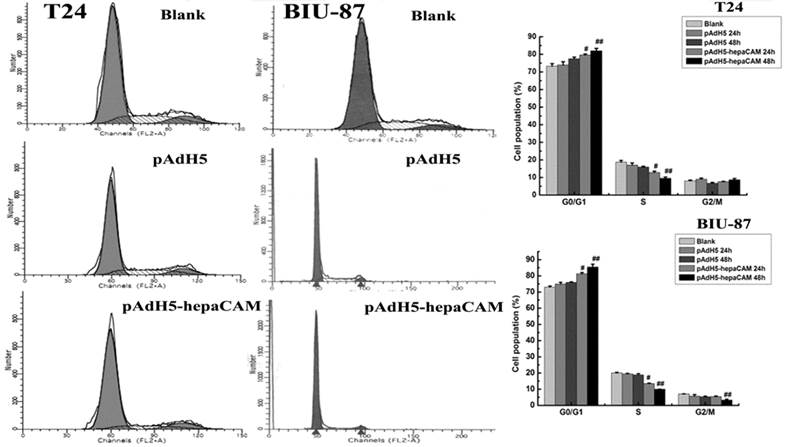
Cell Cycle distribution of T24 and BIU-87 cell lines by FCM.
| Groups | G0/G1 | S | G2/M |
|---|---|---|---|
| blank | a)73.27±1.45 | 18.72±0.99 | 8.01±0.49 |
| b)72.98±0.64 | 19.97±0.53 | 7.04±0.14 | |
| pAdH5 | a)73.97±1.78 | 17.04±1.21 | 8.98±0.62 |
| b)74.93±1.03 | 19.40±0.38 | 5.67±0.94 | |
| pAdH5-hepaCAM | a)81.93±1.48# | 9.57±0.66# | 8.62±0.78 |
| b)85.40±1.80# | 10.03±0.07# | 3.28±0.54 |
a):T24 cell; b): BIU-87 cell; #: p<0.05.
The promoter sequence of hepaCAM
Prediction of hepaCAM promoter showed that promoter sequence is from 14857 to15426. There are 570bp in all. One CpG island was found in promoter sequence by Methprimer (Fig.2A). The blue region was CpG islands and the red thin lines are CpG sites.
Prediction of CpG island in hepaCAM promoter and methylation status of hepaCAM promoter. (A) Promoter prediction of hepaCAM gene was performed by FirstEF software and the results showed that promoter of hepaCAM is from 14857 to15426 of gene sequence. CpG island of hepaCAM promoter was predicted using internet software, the blue region is CpG islands and the red thin lines are CpG sites; (B) MSP used to detect methylation status of hepaCAM gene in bladder cancer cell lines. In blank group, hepaCAM promoter was hypermethylated in T24 and BIU-87 cells, but treated with different concentration of 5-aza-CdR which can reverse the abnormal methylation. 3uM of 5-aza-CdR can significantly repress the hypermethylation of hepaCAM gene in T24 cell line and 5uM for BIU-87 cell line. Meantime, different time of treatment also influence the effect of 5-aza-CdR. In two cells, the perfect time was 72h. (M: Methylation; U: Umethylation; A: 5-aza-CdR).
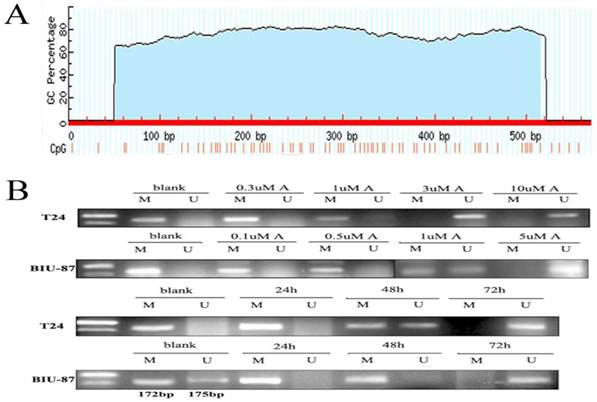
Methylation status of hepaCAM promoter in cells and demethylation of 5-aza-CdR
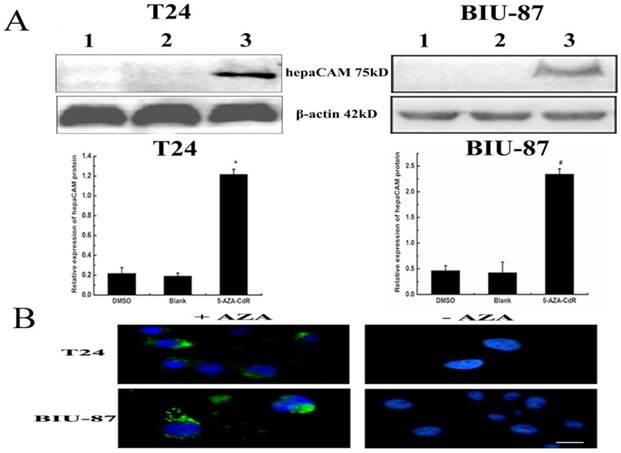
Hypermethylation of hepaCAM promoter was apparently observed in both of T24 and BIU-87 cell lines. Besides, hepaCAM promoter was obviously demethylated by 3uM of 5-Aza-CdR in T24 cell line, and 5uM of 5-Aza-CdR in BIU-87 cell line for 72h (Fig.2B). Demethylation of hepaCAM promoter was not apparent in other concentrations. The expression of hepaCAM mRNA could be restored by 5-Aza-CdR in T24 and BIU-87 cell lines that described before [15]. It was also detected by western-blot (Fig.3A) and Immunofluorescence (Fig.3B). The results revealed that hepaCAM protein was expressed after 5-Aza-CdR treatment. DNA methyltransferases inhibitor 5-Aza-CdR not only reverse the expression of hepaCAM mRNA but also its protein.
5-aza-CdR reverse expression of hepaCAM protein. (A)After T24 and BIU-87 cells treated with 5-aza-CdR,total protein extracted from each group and 120ug of cell lysate was analyzed by Western-blot. The relative intensities of target bands were scanned by Quantity one software and normalized toβ-actin levels. *: p<0.01, #: p<0.05. The results showed that expression of hepaCAM protein was upregulated after 5-aza-CdR treatment in T24 and BIU-87 cell lines (1: DMSO; 2: Blank; 3: 5-aza-CdR.); (B) Immunofluorescence analyzed the re-expression of hepaCAM protein. Immunofluorescence demonstrated that hepaCAM protein re-expressed in T24 and BIU-87 cell lines with 5-aza-CdR treatment (1: cells with 5-aza-CdR treatment; 2: cells without 5-aza-CdR treatment). Image was obtained using Nikon Microscrope. Scale bar= 10 microns.
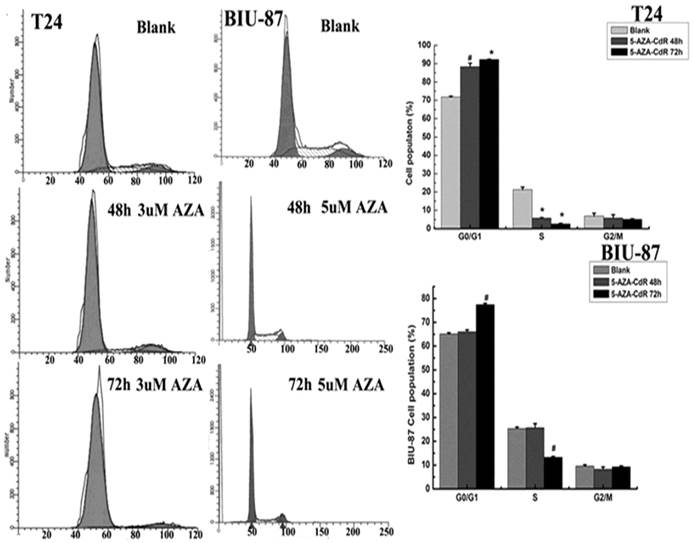
Effect of 5-aza-CdR on cell cycle distribution
According to previous researches, 5-Aza-CdR can re-activate the expression of TSGs, and has been used for treatment of certain leukemia. So the anti-tumor growth of 5-Aza-CdR in bladder cancer cells was analyzed using FCM. The results showed that cell cycle was arrested at G0/G1 after 5-aza-CdR treatment (Fig.4). Cells with 5-aza-CdR treatment were more statistically significant difference than blank cells and DMSO cells (p<0.05) (Table 3).
Effect of 5-aza-CdR on cell cycle distribution in T24 and BIU-87 cells detected by FCM.
| Groups | G0/G1 | S | G2/M |
|---|---|---|---|
| Blank | a)71.79±0.44 | 21.34±1.36 | 6.87±1.53 |
| b)65.10±0.55 | 25.29±0.68 | 9.61±0.52 | |
| 5-aza-CdR/48h | a)88.28±1.96* | 5.85±0.22* | 5.87±1.74 |
| b)66.01±0.80 | 25.69±1.74 | 8.30±0.94 | |
| 5-aza-CdR/72h | a)92.28±0.17# | 2.60±0.37# | 5.12±0.24 |
| b)77.39±0.61# | 13.29±0.36# | 9.32±0.32 |
a):T24 cell; b): BIU-87 cell; #: p<0.01;*:p<0.05.
Effect of 5-aza-CdR on cell cycle distribution. Cells treated with 5-aza-CdR for different time and the cell percentage in each phase of three group were measured by flow cytometer. Data are reported as mean±SD (n=3). The results showed that 5-aza-CdR can arrest cell cycle at G0/G1. The effect of 5-aza-CdR on cell cycle distribution was much apparent at 72h, #: p<0.05, *: p<0.01.
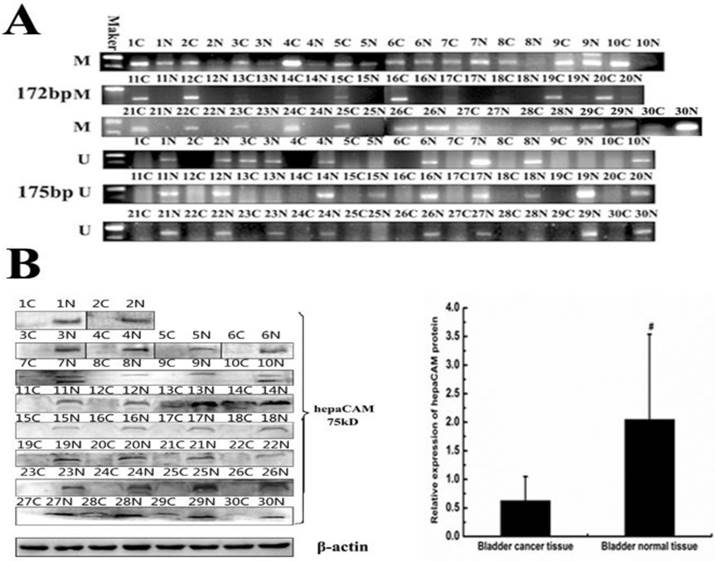
The methylation status of hepaCAM gene promoter and its expression of protein in paired tissues
In vitro, hypermethylation of hepaCAM gene promoter was discovered in cell lines. The matter of wheather the same circumstance occurs in patients with bladder carcinoma is unknown. So methylation status of hepaCAM gene promoter was detected by MSP in bladder cancer tissues (Fig.5A). The positive rate of hepaCAM promoter methylation in tumor tissues was 83.33%, which was obviously higher than that in adjacent normal bladder tissues (33.3%, p<0.05). Results of Western-blot revealed that hepaCAM protein mostly expressed in adjacent normal tissues of patients (Fig.5B). It expressed in only few tumor tissues (p<0.01). Spearman correlation analysis demonstrated a negative correlation between expression of hepaCAM protein and its promoter hypermethylation (correlation coefficient r=-0.894, p=0.000) (Table 4).
The correlation between expression of hepaCAM protein and hepaCAM promoter methylaion by MSP.
| parameters | methylation | |
|---|---|---|
| + | - | |
| Deletion of hepaCAM protein | 24 | 0 |
| No change of hepaCAM protein | 1 | 5 |
| r | -0.894 | |
| p* | 0.000 | |
*: p<0.01 was considered statistically significant.
Methylation status of hepaCAM gene and expression of hepaCAM protein in bladder cancer tissue specimens. (A) Genome DNA were extracted and modified with treatment of sodium bisulfite. Modified DNA was used as template for MSP to detect methylation status of hepaCAM promoter in 30 paires patients. Results showed that 25 of 30 (83.3%) patients' tumor tissues occurs hypermethylation of hepaCAM promoter, and 10 of 30 (33.3%) patients' adjacent normal tissues occurs hypermethylation ( C: cancer tissue; N: normal tissue in side of cancer; M: methylation; U: unmethylation). (B)Total protein extracted from tissue specimens. Western-blot performed as described above to detect the expression of hepaCAM protein in 30 paires patients. In most of bladder cancer tissues, the expression of hepaCAM protein was lost, and it expressed in corresponding adjacent normal tissues. #: p<0.01(C: cancer tissue; N: normal tissue in adjacent cancer).
Discussion
Bladder cancer is the most common cancer in urologic neoplasms in China, and its incidence ranked 8th in systemic malignancy[16]. Bladder transitional cell carcinoma accounted for 90% of bladder tumor[17] and its biological behavior is complicated and changeable, with high heterogeneity and recurrent. The "gold standard" of current clinical diagnosis is cystoscopy with biopsy. This method is traumatic and difficult for patients with invisible tumors.
Methylation of DNA is a chemical modification process. The methyl group of S-adenosylmethionine transfers to the carbon five prime position of a cytosine pyrimidine ring of DNA under the action of DNA methyltransferase (DNMT), forming 5-methylcytosine. DNA methylation is an important mechanism for the inactivation of malignant tumor suppressor genes and occurs mainly in CpG islands[18]. Methylation of CpG islands in suppressor genes promoter occurs in most tumors. Moreover, abnormal DNA methylation occurred in early tumorigenesis, contributing as an ideal marker for early diagnosis of tumors[19]. Some studies have proved that epigenetic changes of multiple TSGs (tumor suppressor genes) were involved in breast pathogenesis[20]. Researchers also demonstrated that aberrant methylated genes played various functions in the cell cycle regulation, proliferation and adhesion[21]. Research of Jelilek J showed that aberrant DNA methylation is associated with CML progression[22]. In addition, Stephen JK' research suggested that abnormal methylation of CASP8, RASSF1, and NIS maybe early changed in thyroid tumorigenesis regardless of cell type[23]. These studies showed that methylation of TSGs promoter played a significant role in tumorigenesis.
As a tumor suppressor gene, the expression of hepaCAM was lost in human hepatocellular carcinoma (HCC)[24-26]. Previous studies also showed that hepaCAM played a similar role in transitional cell carcinoma of the bladder (TCCB) as well as in HCC[26]. To confirm that hypermethylation of hepaCAM promoter leads to its inactivation in bladder carcinoma, we analyzed the methylation status of hepaCAM promoter in bladder cancer cells and tumor tissues. We found a CpG island in hepaCAM promoter through Internet software (http://rulai.cshl.org/tools/FirstEF/ and http://www.urogene.org/methprimer/index1.html). Hypermethylation of hepaCAM promoter was detected by MSP in bladder cancer cells (T24 and BIU-87) and in 83.3% of patients. The expression of hepaCAM protein was lost in 80% patients. These results demonstrated that inactivation of hepaCAM gene may be caused by hypermethylation of promoter. Previous report[19]demonstrated that the methylation of hepaCAM gene was occurred at exon 2. Whether the methylation of hepaCAM gene promoter and exon 2 were the conjunct mechanism of gene silencing need in-depth investigate.
To further identify this, T24 and BIU-87 cell lines were treated with different concentration of 5-aza-CdR to investigate the effect of 5-aza-CdR on the expression of hepaCAM mRNA, protein and promoter methylation. 5-aza-CdR covalently binds DNA methyltransferase, which then become trapped and unable to catalyse the methylation of newly synthesized DNA strands[27]. Clinical trials found that DNA methylation gradually disappears in the tumor cells from responders with 5-Aza-CdR treatment[27]. 5-aza-CdR is phosphorylated by deoxycytidine kinase, and its end product, decitabine triphosphate, is incorporated into DNA[28]. Incorporation of a high concentration of decitabine triphosphate into DNA can inhibit DNA synthesis and induce cell cycle arrest[29-31]. 5-aza-CdR has been used in clinical phase III trials patients with Myelodysplastic syndrome (MDS)[28]. Ju Hee Kim's research showed that 5-Aza-CdR can reverse expression of CAMK2B and ARFGEF1, besides hypermethylation of CAMK2B and ARFGEF1can inhibit gene activity in cancer cells[32]. Trenton et al. showed that 5-aza-CdR significantly increased the expression of PHD3[33]. Previous studies demonstrated that treatment of 5-Aza-CdR in corresponding concentration for 72h in T24 and BIU-87 cell lines can reverse the expression of hepaCAM mRNA[15]. These studies revealed that 5-Aza-CdR can also reverse the expression of hepaCAM protein. This drug affects the cell cycle distribution, arresting the cell cycle at G0/G1 phase. It also shows that 5-aza-CdR can restrain bladder cancer cells' growth and reverse the hypermethylation of hepaCAM promoter.
In conclusion, hypermethylation occurs in hepaCAM promoter region. Demethylating drug (5-aza-CdR) can reverse its expression of mRNA and protein, and inhibit the growth of tumor cells. Hypermethylation of promoters is a major gene silencing mechanisms of hepaCAM in human bladder cancer. Further research is required to identify that which promoter sites mostly affects transcriptional level.
Abbreviations
hepaCAM, hepatocyte cell adhesion molecule; 5-aza-CdR, 5-Aza-2'-deoxycytidine; TGSs, tumor suppressor genes; MSP, Methylation-specific polymerase chain reaction; FCM, Flow Cytometry
Acknowledgements
This work was supported by National Natural Science Foundation of P.R. China (Grant No. 81072086).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chung Moh M, Hoon Lee L, Shen S. et al. Cloning and characterization of hepaCAM, a novel Ig-like cell adhesion molecule suppressed in human hepatocellular carcinoma. J Hepatol. 2005;42(6):833-41
2. He Y, Wu X, Luo C. et al. Functional significance of the hepaCAM gene in bladder cancer. BMC Cancer. 2010Mar8;10:83
3. Yang S, Wu X, Luo C. et al. Expression and clinical significance of hepaCAM and VEGF in urothelial carcinoma. World J Urol. 2010;28(4):473-8
4. Partha M. Das, Rakesh Singal.DNA Methylation and Cancer. J Clin Oncol. 2004;22:4632-4642
5. Adrian Bird. DNA methylation patterns and epigenetic memory. GENES & DEVELOPMENT. 2012;16:6-21
6. Ahmed IA, Pusch CM, Hamed T. et al. Epigenetic alterations by methylation of RASSF1A and DAPK1 promoter sequences in mammary carcinoma detected in extracellular tumor DNA. Cancer Genet Cytogenet. 2010;199(2):96-100
7. Wang S, Xiao X, Zhou X. et al. TFPI-2 is a putative tumor suppressor gene frequently inactivated by promoter hypermethylation in nasopharyngeal carcinoma. BMC Cancer. 2010;9:10-617
8. Barzily-Rokni M, Friedman N, Ron-Bigger S. et al. Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A). Nucleic Acids Res. 2011;39(4):1326-35
9. Caldeira JR, Prando EC, Quevedo FC. et al. CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer. 2006;6:48
10. Jeong-Seok Nam, Yoshinori Ino, Yae Kanai. et al. 5-Aza-2-deoxycytidine restores the E-cadherin system in E-cadherin-silenced cancer cells and reduces cancer metastasis. Clinical & Experimental Metastasis. 2004;21:49-56
11. Nuno J. Farinha, Sepideh Shaker, Maryse Lemaire, et al. Momparler. Activation of Expression of p15, p73 and E-Cadherin in Leukemic Cells by Different Concentrations of 5-Aza-2'-Deoxycytidine (Decitabine). ANTICANCER RESEARCH. 2004;24:75-78
12. Pinto A. & Zagonel, V. Leukemia. 1993;7:51-60
13. Ramana V. Davuluri, Ivo Grosse, Michael Q. Zhang. Computational identification of promoters and first exons in the human genome. Nature Genetics. 2001;29:412-417
14. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427-31
15. Pan C, Wu X, Luo C. et al. Exon 2 methylation inhibits hepaCAM expression in transitional cell carcinoma of the bladder. Urol Int. 2010;85(3):347-54
16. Babjuk MO, Osterlinck W, Sylvester R. et al. EAU guidelines on-nonmuscle -carcinoma of the bladder. EAU guidelines. 2009;33(4):36l-371
17. Oosterlinck W, Lobel B, Jakse G. et al. Guidelines on bladder cancer. Eur Urol. 2002;41:105-112
18. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415-428
19. Esteller M, Corn PG. Agene hypermethylation profile of human cancer. Cancer research. 2001;61(8):3225-29
20. Xiang TX, Yuan Y, Li LL. et al. Aberrant promoter CpG methylation and its translational applications in breast cancer. Chin J Cancer. 2011 doi: 10.5732/cjc.011.10344
21. Marzese DM, Gago FE, Orozco JI. et al. Aberrant DNA methylation of cancer -related genes in giant breast fibroadenoma:a case report[J]. J Med Case Reports. 2011;5(1):516
22. Jelinek J, Gharibyan V, Estecio MR. et al. Aberrant DNA methylation is associated with disease progression, resistance to imatinib andshortened survival in chronic myelogenous leukemia. PLoS One. 2011;6(7):e22110
23. Stephen JK, Chitale D, Narra V. et al. DNA methylation in thyroid tumorigenesis. Cancers (Basel). 2011;3(2):1732-1743
24. Moh MC, Zhang C, Luo C. et al. Structural and functional analyses of a novel ig-like cell adhesion molecule, hepaCAM, in the human breast carcinoma MCF7 cells. J Biol Chem. 2005;280(29):27366-27374
25. Lee LH, Moh MC, Zhang T. et al. The immunoglobulin-like cell adhesion molecule hepaCAM induces differentiation of human glioblastoma U373-MG cells. J Cell Biochem. 2009;15:1129-1138
26. Moh MC, Zhang T, Lee LH. et al. Expression of hepaCAM is downregulated in cancers and induces senescence-like growth arrest via a p53/p21-dependent pathway in human breast cancer cells. Carcinogenesis. 2008;29(12):2298-2305
27. Grønbaek K, Treppendahl M, Asmar F. et al. Epigenetic changes in cancer as potential targets for prophylaxis and maintenance therapy. Basic Clin Pharmacol Toxicol. 2008;03(5):389-96
28. Oki Y, Issa JP. Recent Clinical Trials in Epigenetic Therapy. Rev Recent Clin Trials. 2006;1(2):169-82
29. Li LH, Olin EJ, Fraser TJ. et al. Phase specificity of 5-azacytidine against mammalian cells in tissue culture. Cancer Res. 1970;30:2770-5
30. Cihak A, Vesely J. Prolongation of the lag period preceding the enhancement of thymidine and thymidylate kinase activity in regenerating rat liver by 5-azacytidine. Biochem Pharmacol. 1972;21:3257-65
31. Cihak A, Vesely J, Skoda J. Azapyrimidine nucleosides: metabolism and inhibitory mechanisms. Adv Enzyme Regul. 1985;24:335-54
32. Kim JH, Kim TW, Kim SJ. Downregulation of ARFGEF1 and CAMK2B by promoter hypermethylation in breast cancer cells. BMB Rep. 2011;44(8):523-8
33. Place TL, Fitzgerald MP, Venkataraman S. et al. Aberrant Promoter CpG Methylation Is a Mechanism for Impaired PHD3 Expression in a Diverse Set of Malignan Cells. PLoS One. 2011;6(1):e14617
Author contact
![]() Corresponding author: Chunli Luo, Department of Laboratory Diagnosis, Chongqing Medical University, No.1, Yixueyuan Road, Yuzhong Restrict, Chongqing, 400016, China. E-mail: luochunli79com. Fax: 023-68485005. Tel: 023-68485223.
Corresponding author: Chunli Luo, Department of Laboratory Diagnosis, Chongqing Medical University, No.1, Yixueyuan Road, Yuzhong Restrict, Chongqing, 400016, China. E-mail: luochunli79com. Fax: 023-68485005. Tel: 023-68485223.