Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(8):938-947. doi:10.7150/ijms.6152 This issue Cite
Research Paper
C-Reactive Protein and Serum Amyloid A Overexpression in Lung Tissues of Chronic Obstructive Pulmonary Disease Patients: A Case-Control Study
Jose Luis López-Campos1,2,3, ![]() , Carmen Calero1,2,3, Belén Rojano1, Marta López-Porras1, Javier Sáenz-Coronilla2, Ana I Blanco1, Verónica Sánchez-López2, Daniela Tobar1, Ana Montes-Worboys2,3*, Elena Arellano2*
, Carmen Calero1,2,3, Belén Rojano1, Marta López-Porras1, Javier Sáenz-Coronilla2, Ana I Blanco1, Verónica Sánchez-López2, Daniela Tobar1, Ana Montes-Worboys2,3*, Elena Arellano2*
1. Unidad Medico-Quirurgica de Enfermedades Respiratorias. Hospital Universitario Virgen del Rocio, Seville, Spain.
2. Instituto de Biomedicina de Sevilla (IBiS). Hospital Universitario Virgen del Rocio, Seville, Spain.
3. CIBER de Enfermedades Respiratorias (CIBERES), Spain
* Both authors have contributed equally to this work.
Received 2013-2-26; Accepted 2013-5-13; Published 2013-6-8
Abstract
Background. Although researchers have consistently demonstrated systemic inflammation in chronic obstructive pulmonary disease (COPD), its origin is yet unknown. We aimed to compare the lung bronchial and parenchymal tissues as potential sources of major acute-phase reactants in COPD patients and resistant smokers.
Methods. Consecutive patients undergoing elective surgery for suspected primary lung cancer were considered for the study. Patients were categorized as COPD or resistant smokers according to their spirometric results. Lung parenchyma and bronchus sections distant from the primary lesion were obtained. C-reactive protein (CRP) and serum amyloid A (SAA1, SAA2 and SAA4) gene expressions were evaluated by RT-PCR. Protein levels were evaluated in paraffin embedded lung tissues by immunohistochemistry and in serum samples by nephelometry.
Results. Our study included 85 patients with COPD and 87 resistant smokers. In bronchial and parenchymal tissues, both CRP and SAA were overexpressed in COPD patients. In the bronchus, CRP, SAA1, SAA2, and SA4 gene expressions in COPD patients were 1.89-fold, 4.36-fold, 3.65-fold, and 3.9-fold the control values, respectively. In the parenchyma, CRP, SAA1, and SAA2 gene expressions were 2.41-, 1.97-, and 1.76-fold the control values, respectively. Immunohistochemistry showed an over-stained pattern of these markers on endovascular cells of COPD patients. There was no correlation with serum protein concentration.
Conclusions. These results indicate an overexpression of CRP and SAA in both bronchial and parenchymal tissue in COPD, which differs between both locations, indicating tissue/cell type specificity. The endothelial cells might play a role in the production of theses markers.
Keywords: COPD, C-reactive protein, Serum Amyloid A, gene expression, immunohistochemistry.
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading cause of morbidity and mortality worldwide, considerably impairing the health-related quality of life. COPD is also associated with systemic effects, among which cardiovascular disease, skeletal muscle dysfunction, and systemic inflammation have been studied in detail [1].
Systemic inflammation in COPD is defined as increased levels of inflammatory markers from different biological pathways [2]. C-reactive protein (CRP) is a major acute-phase reactant and one of the more deeply studied molecules of the human body in relation with COPD [3]. Serum amyloid A (SAA), another major acute-phase reactant, is also associated with COPD [4]. Interestingly, CRP and SAA share secretory stimuli, with a similar increase pattern in the serum [5]. Although these inflammatory markers show consistent increase, issues such as wide variations in these elevations among COPD patients or prognostic cut-off values remain to be addressed.
The most obvious explanation for the presence of systemic inflammation in these patients is that this pulmonary inflammation somehow “spills over” into the systemic circulation [6]; however, the results of previous studies do not completely support this hypothesis. Although proteins originating from the lung may exert systemic effects, there is a lack of correlation between airway cytokine concentrations and those in the circulation [7], and investigators have been unable to find an association between the inflammatory load of induced sputum and plasma.
To verify the spill over hypothesis investigation should start to address whether lung tissues can produce these biomarkers in COPD. Previous studies have described that lung tissues can synthesize acute-phase biomarkers in normal tissues and in animal or cell models [8-10]. However, it has not yet been investigated whether lung tissues can synthesize inflammatory mediators of COPD in comparison with non-COPD (resistant) smokers. The closest study was recently published showing a non-specific immunohistochemistry staining for SAA in macrophages close to the airway epithelium of COPD patients [11]. Interestingly, studies have evaluated protein production or gene expression in bronchial tissue or lung parenchyma without comparing the results between these compartments. Tissue specificity in different respiratory system compartments in COPD as compared to resistant smokers has not yet been investigated.
In the present study we aimed to analyse the lung tissues production of major acute-phase reactants, evaluate the site of production and correlate with the levels of the same biomarkers in serum samples. We conducted a case-control design to evaluate COPD patients and non-COPD smokers who underwent resection of suspected primary lung neoplasm. We analysed the gene expression of CRP and SAA using reverse transcriptase-polymerase chain reaction (RT-PCR) in both bronchial tissue and lung parenchyma. Additionally, we evaluated the tissue protein production by immunohistochemistry and serum protein concentration by nephelometry. Thus, we were able to assess if the production of these acute-phase reactants is different in both locations, provide information on the location of this overproduction, and if it correlates with the systemic inflammatory load as measured by these two biomarkers.
Methods
Subjects
We recruited consecutive patients in the surgical waiting list who were about to undergo elective pneumectomy or lobectomy for suspected primary lung cancer from February 2008 to June 2011. The study was approved by the Institutional Review Board from Hospital Virgen del Rocío, and patients provided written informed consent prior to their inclusion in the study. The patients were identified upon the day of admission, i.e. a day before surgery was planned. Patients who were <40 years of age, had an acute respiratory infection during the previous 2 months, had a previously diagnosed neoplasm, received radiotherapy or chemotherapy, or suffered from chronic inflammatory diseases were excluded from the study. Further, the time spent from the opening of the cutaneous layer through the extraction of the anatomical sample was measured, and cases in which this time was >3 hours were also excluded from the study, since any potential stimulation of the studied biomarkers due to surgery cannot be ruled out.
Medical records were checked to ensure that the patients had a recent lung function test. Those whose spirometry results revealed a forced expiratory volume in the first second (FEV1)/forced vital capacity (FVC) ratio < 0.7 were considered in the COPD group, and the remaining resistant smokers were used as the control subjects. All patients completed a standardized questionnaire recording their medical history, tobacco consumption, and actual treatments. Further, the TNM staging of the primary lesion [12] and the surgical procedure data were also collected. During the surgery, a sample within the resected bronchus was taken for microbiological studies prior to the resection in order to evaluate microbiological colonization. Additionally, the microbiological samples of the bronchoscopic studies prior to the surgery were also reviewed.
After the surgery, a 5-10-g section of lung parenchyma and the largest available section of the resected bronchus were selected. Samples that were the most distally located from the primary lesion were immediately processed in our laboratory. The rest of the anatomical sample was sent to the pathology department for the diagnosis of the primary lesion.
Tissue processing
Tissue samples were analyzed using quantitative RT-PCR, which consists of 3 phases: RNA extraction, reverse transcription to cDNA, and gene amplification. RNA was isolated from fresh tissue following the TriSure manufacturer's protocol (Bioline, London, UK). Then, RNA was treated with DNAase free of RNase using a commercial kit (QIAgen, GmbH) to remove any residual genomic DNA, and cDNA was synthesized using an iScript kit (Bio-Rad, CA). Each reaction was duplicated at a total volume of 25 µl and contained 2 µl cDNA (40 ng/µl), 12.5 µl SYBR Green PCR master mix (Stratagene, CA) and 10.5 µl primers/H2O. RT-qPCR was performed using a MX3005P system (Stratagene) at 95ºC for 30 s, 60ºC for 1 min, and 72ºC for 30 s. Gene amplification was normalized to 18s RNA. The primers used for amplification are described in Table 1. Since human SAA protein consists of 3 tightly linked genes (SAA1, SAA2 and SAA4) [13], the GeneBank database from the National Center for Biotechnology Information (NCBI) was consulted and those portions of the genes which were not in the homology region were selected. These primers were then synthesized ad hoc by an external company (Sigma-Aldrich).
Primers used.
| Forward | Reverse | |
|---|---|---|
| 18s | TGAAATATCCAGAACATCTTA | GCAAAATTTATTGTCCCATCAT |
| CRP | GTGTTTCCCAAAGAGTCGGATA | CCACGGGTCGAGGACAGTT |
| SAA1 | ATCAGCGATGCCAGAGAGAAT | GTGATTGGGGTCTTTGCCA |
| SAA2 | AGCCAATTACATCGGCTCAG | ATTTATTGGCAGCCTGATCG |
| SAA4 | GTCCAACGAGAAAGCTGAGG | AGTGACCCTGTGTCCCTGTC |
Serum analysis
Serum CRP and SAA were measured by immunonephelometry (Dade Behring, Marbrug, Germany) according to the manufacturer's instructions. The nephelometric determinations of CRP were performed using a polystyrene-enhanced immunonephelometric method on a Dimension Vista System (Siemens, Munich, Germany). Commercially available kits were used (Dade Behring, Marburg, Germany). The nephelometric determinations of SAA were performed using a latex-enhanced immunonephelometric method on a Dade Behring BN2 Nephelometer Analyzer equipped with commercially available kits (Dade Behring, Marburg, Germany).
Immunohistochemistry
Tissue blocks from the subpleural parenchyma avoiding areas involved by tumor were fixed in 10% formalin, embedded in paraffin, and 5-µm sections were prepared for immunohistochemical analysis. Mouse monoclonal antibodies against SAA1 (Novus Biologicals, Cambridge UK), and CRP (Abcam, Cambridge UK) were used. Antigen retrieval was achieved by microwave heat treatment in citrate buffer (Dako, USA) at 98ºC for 15 min. Bound antibody was developed with daiminobenzidine using a Dako Envision staining kit (K4065) according to manufacturer´s recommendations. Stained sections were observed under light microscope by two independent observers. All immunohistochemical studies were performed including the standard quality controls. As a negative control, we performed an immunohistochemical study using secondary antibodies without the primary antibody.
Statistical analysis
Statistical computations were performed using the Statistical Package for Social Sciences (SPSS, IBM Corporation. Somers, NY) version 20.0. Clinical variables were presented as the mean with standard deviations or the absolute and relative frequencies depending on the nature of the variable. Laboratory data on gene expression were analyzed using the 2-∆∆Ct method [14]. Comparisons of gene expression between both anatomical locations were performed using the Wilcoxon test, and comparisons between cases and controls were evaluated by the Mann-Whitney test. Simple linear correlations between tissue gene expression and serum protein concentration were studied by Spearman coefficient. Alpha error was set at 0.05.
Results
Patients and procedures
Our study included 85 patients with COPD (95% males, age: 67 ± 7 years, FEV1 69.5 ± 17.2%) and 87 resistant smokers. The clinical characteristics of the patients are summarised in Table 2. The patients underwent pneumectomies (9.3%), lobectomies (80.2%), and atypical resections (10.5%). Interventions were performed on the right and left hemithorax in 60.2% and 39.8% of the patients, respectively. Adenocarcinomas (36.6%) and squamous cell carcinoma (34.3%) were the most frequently encountered types of cancer. After the revision of histology, 6.4% of the cases had benign tumors and 8.1% of the cases had non-neoplastic disorders, which accounted for 15.1% of non-malignant tumors. Most of the malignant cases (41.8%) were stage I subtypes. COPD patients had malignant histology more frequently (94.1% vs. 75.9%, p = 0.001). Five cases in each group resulted to have a positive culture either in the bronchoscopic or in the surgical samples.
Characteristics of the patients.
| Controls (n=87) | COPD (n=85) | P value* | |
|---|---|---|---|
| Males (n) | 62 (71.3%) | 81 (95.3%) | < 0.001 |
| Age (years) | 61.4 (11.8) | 67.6 (7.5) | < 0.001 |
| Tobacco history (pack-years) | 37.6 (32.4) | 63.2 (35.3) | < 0.001 |
| GOLD stage: | |||
| - GOLD 1 | 23 (27.7%) | ||
| - GOLD 2 | 52 (62.7%) | ||
| - GOLD 3 | 8 (9.6%) | ||
| - Not available† | 2 (2.3%) | ||
| Charlson-age index | 4.2 (2.2) | 5.9 (1.5) | < 0.001 |
| Inhaled corticosteroids use | - | 16 (18.8%) | |
| Long-acting ß2 agonists use | - | 17 (20%) | |
| Tiotropium use | - | 27 (31.8%) | |
| FVC (%) | 102.7 (79.7) | 91.3 (19.7) | NS |
| FEV1 (%) | 89.1 (16.9) | 69.5 (17.2) | < 0.001 |
| FEV1/FVC (%) | 77.5 (7.9) | 59.0 (8.1) | < 0.001 |
| Patients taking statins (n) | 18 (20.7) | 25 (29.4) | NS |
Data expressed as mean (standard deviation) or absolute (relative) frequencies as requested. * p value calculated by χ2 test or Student t test for unpaired data as appropriate. NS: not significant. †Two COPD patients could not provide recent spirometric data: One tracheostomised and one could not collaborate. Both had old spirometric values confirming the diagnosis of COPD.
Acute-phase reactants in bronchial tissue
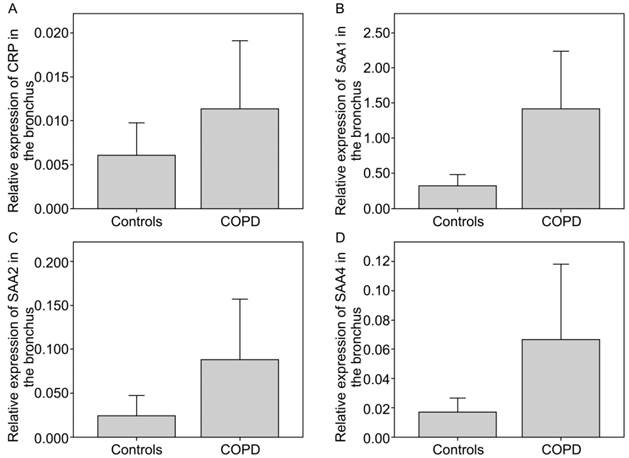
CRP and SAA were overexpressed in bronchial tissue in patients with COPD as compared to the controls (Figure 1a). CRP expression was 1.89-fold higher in COPD patients than in the controls. Further, SAA expression was higher in bronchial tissue (Figure 1b-d). The SAA1, SAA2, and SAA4 expressions in COPD patients increased by a 4.36-fold (p=0.013), 3.65-fold (p=0.004), and 3.9-fold the control values, respectively. The biomarker expressions in patients with lung malignancy were not different in patients taking statins or in those with non-neoplastic diseases in both COPD and controls (data not shown).
Relative gene expression of CRP and SAA normalised with 18s RNA in COPD vs. controls in the bronchus. a) CRP, b)SAA1 (p = 0.013), c) SAA2 (p = 0.004), d) SAA4.
Expression of acute-phase reactants in parenchymal tissue
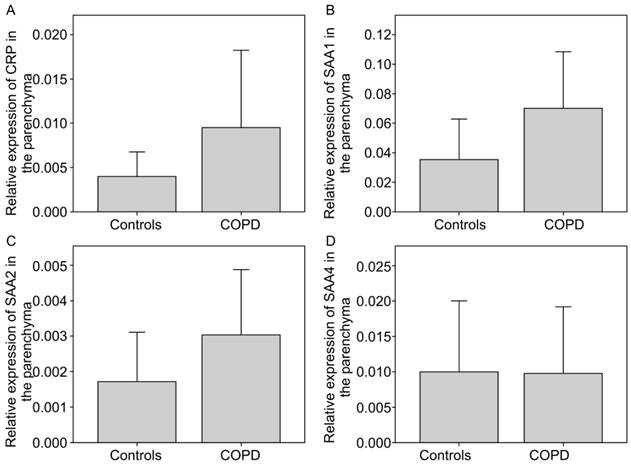
CRP and SAA isoforms were found to be overexpressed in COPD as compared to the controls (Figure 2a). CRP expression was 2.41-fold higher in COPD than in the controls. Further, SAA expression was increased in bronchial tissue in COPD (Figure 2b-d). SAA1 and SAA2 in COPD showed a mean increase of 1.97-fold and 1.76-fold (p=0.039) of the control, respectively. SAA4 of COPD and controls showed no difference. Patients taking statins or those with lung malignancy did not show different expressions as compared to those with non-neoplastic disease for both COPD and controls (data not shown).
Relative gene expression of CRP and SAA normalised with 18s RNA in COPD vs. controls in the parenchyma. a) CRP, b)SAA1, c) SAA2 (p = 0.039), d) SAA4.
Expression of bronchial and parenchymal tissues
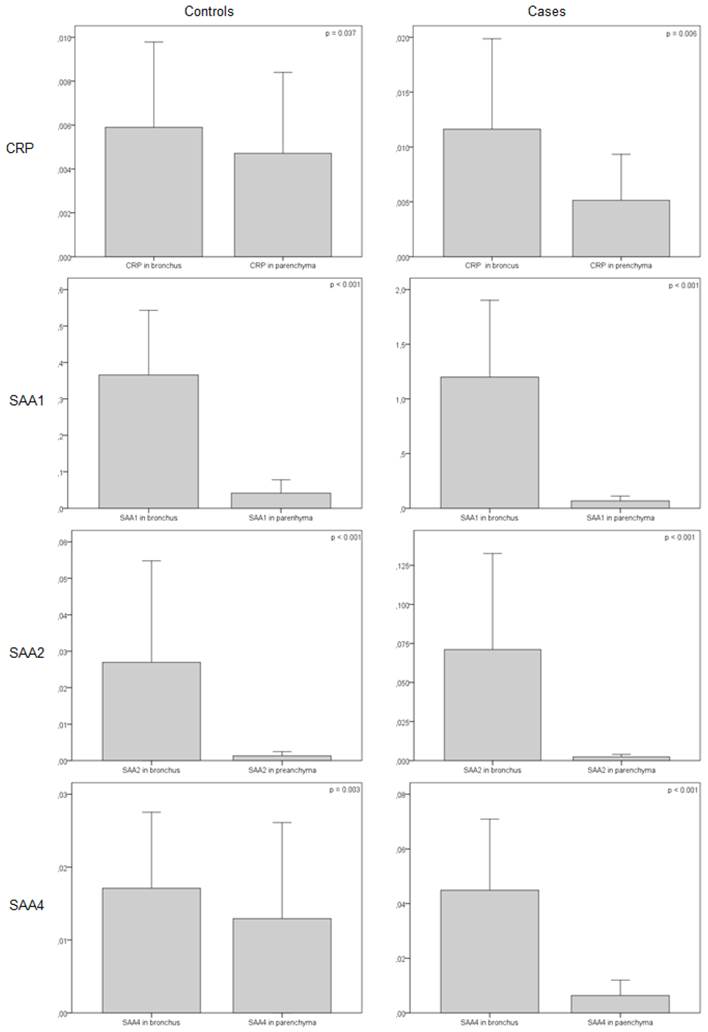
Interestingly, when the marker expressions were compared in the bronchus and parenchyma, the former expressed more acute-phase reactants in both COPD and control subjects. This expression pattern was also higher in COPD than in controls (Figure 3).
Relative gene expression of CRP and SAA normalised with 18s RNA in COPD vs. controls (bronchus vs. parenchyma).
Correlation with serum protein concentration
The levels of serum CRP in COPD patients were higher as compared to controls (22.5 (34.5) vs 7.5 (12.5); p = 0.016). The levels of SAA were also elevated in serum of COPD patients (81.5 (225.6) vs 12.7 (37.5), p = 0.002). However, no significant correlation was found between lung tissues gene expression and serum protein concentration (data not shown).
Immunohistochemistry
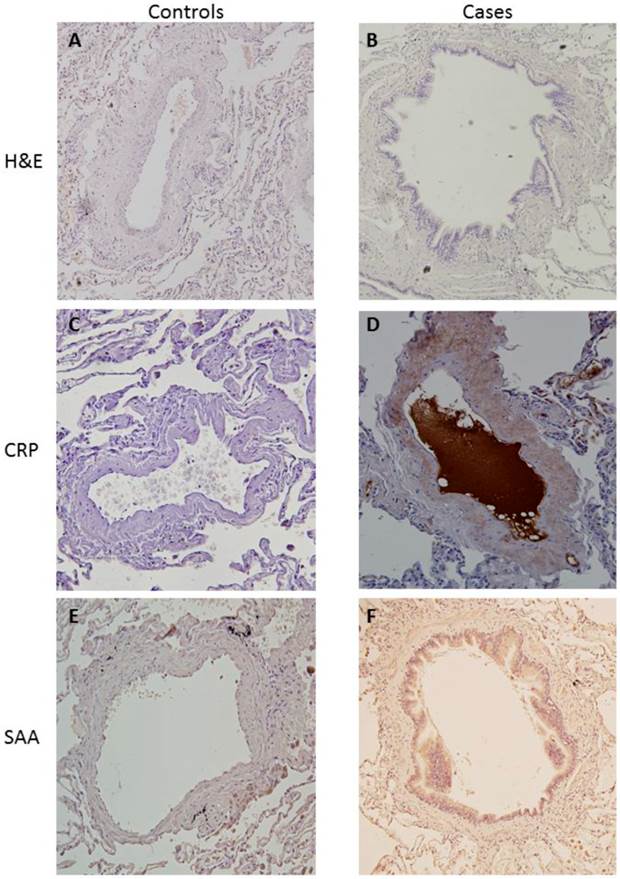
Immunohistochemistry analysis showed an increased expression of both CRP and SAA in lung tissue in COPD as compared to controls. As shown in figure 4, staining was captured by vessel wall most importantly. We also observed an unspecific increased staining in macrophages.
Immunohistochemistry for cases and controls. a) H&E for controls. b) H&E for cases. c) PCR staining for controls. d) PCR staining for cases. e) SAA1 staining for controls. f) SAA1 staining for cases.
Discussion
This study provides novel data on CRP and SAA synthesis in lung parenchyma and bronchial tissue in patients with COPD and resistant smokers. Our results indicate that both parenchyma and bronchus synthesize CRP and SAA de novo and that their production is enhanced in COPD. Further, the production profiles differed between the parenchyma and bronchus in both groups, suggesting tissue or cell specificity. Additionally, our findings suggest that the lung vasculature is one of the possible sites of production. However, we fail to demonstrate that this overexpression is related to serum protein concentration.
In the present study we have evaluated whole tissue samples. Previous studies have demonstrated local production of SAA proteins in histologically normal human lung tissues rich in endothelial cells and macrophages, which express SAA [8]. SAA proteins consist of 3 tightly linked genes (SAA1, SAA2, and SAA4) acting as apolipoproteins and synthesized in response to cytokines released by activated monocytes/macrophages [13]. The overall gene sequences of SAA1 and SAA2 are approximately 95% identical, whereas the mature SAA4 protein shares only 55% identity with human SAA1 and SAA2 [13]. The SAA4 locus contains the gene encoding for a unique SAA family member, which is constitutively expressed on high density lipoprotein (HDL) [15], whereas SAA1 and SAA2 constitute acute SAA. SAA production has been described in smooth muscle aortic cells [16] and atherosclerotic lesions [17], as well as in macrophages from sarcoid granulomas [18]. Interestingly, SAA has also been described in lung macrophages close to the airway epithelium of COPD patients [11]. Similarly, CRP has been found to be elevated in bronchial tissues and bronchial epithelial cell models [9, 10]. Interestingly, CRP is also produced by alveolar macrophages [19]. Thus, there are many different cell types that may be responsible for this production in COPD. Our findings suggest that the lung vasculature is one of the main sites of production which would be in accordance with current knowledge. The overproduction of acute phase reactants by the vessel wall is not new. Aortic endothelial cells have been described as a source of CRP with proinflammatory effects [20] and SAA has been found in different locations in endothelial cells [21]. However, no studies had assessed the vessel wall as the source of systemic inflammation in COPD so far.
The differential expression of biomarkers depending on their location is remarkable. Although COPD affects the entire respiratory system, the implication of the different compartments (airway, lung parenchyma, and pulmonary vasculature) varies from patient to patient, confirming the presence of disease phenotypes [22, 23]. Interestingly, the different degrees of intervention of airway and lung parenchyma in the pathogenesis of COPD have been recently described [24]. Therefore, it is possible that biomarker production may differ according to different disease phenotypes [25]. However, categorising patient into well-defined clinical phenotypes is still a matter of controversy [26, 27]. Our results provide some information on the biomarkers production in different location which opens a new area for research in this field.
The mechanisms driving CRP and SAA lung expression are unknown. Among those factors known to initiate and maintain inflammation, there are several hormones that regulate these biomarkers expression. These include interleukin (IL)-1, IL-6, IL-11, glucocorticoids, oncostatin M, leukemia inhibitory factor, tumor necrosis factor α, transforming growth factor β, interferon γ, ciliary neurotrophic factor and retinoid acid [28], many of which have a key role in the pathogenesis of COPD. So it is possible that either a local stimulus may enhance this overproduction in COPD or that this is due to a systemic response. Future studies will have to evaluate the potential underlying mechanisms.
The “spill over” hypothesis indicates the potential role of the lung tissues in the systemic inflammatory load [6]. Although proteins originating from the lung may exert systemic effects, there is a lack of correlation between airway cytokine concentrations and those in the circulation and previous studies have also failed to demonstrate this association [7]. In this regard, there are a few aspects that merit consideration. Firstly, although a correlation between serum and tissue expression should be obtained to support the idea behind the spill over hypothesis, the detection of the presence of mRNA provides no information on whether that mRNA will be translated into a protein or whether a functional protein will be translated [29]. In this regard, there are several well-known mechanisms that may interact with the implicated biological pathways. In fact, it is now well established that there is frequently a lack of concordance between mRNA and protein concentration data [30]. Secondly, the present study and the previous ones are cross-sectional in design. This type of study provides valuable information on a particular time point. However, COPD is a constantly evolving disease and longitudinal data would be probably needed to thoroughly assess the relationship between local production and the systemic consequence. Finally, another possibility to explain this lack of correlation may be the different involvement of the lung vasculature depending on the studied lobe or the disease phenotype. In this regard, more or less emphysematous tissue or airway wall thickness in one particular individual may exert an influence on the vasculature studied and contribute differently to systemic inflammation. Interestingly, this systemic inflammation has been described to be very variable between studied subjects [31]. In this regard, systemic vasculature has not been deeply studied as a potential source of systemic inflammation in COPD, and some recent information is already available [32]. In view of overcoming these lacunas future steps should include longitudinal association studies between the overproduction of these biomarkers in the pulmonary or systemic vasculature and systemic inflammation.
Although all cases were operated for suspected malignancy, 15.1% did not have malignant disease. COPD patients were more likely to have a malignancy. Although this was not our study aim, our finding is not surprising. Several studies have addressed the relationship between these two diseases, which are linked together by more than the common risk factor of tobacco smoke—both processes share several genetic and epigenetic markers [33-35]. However, with respect to local inflammation, we were unable to find an association between lung malignancy and the gene expression of these biomarkers.
In summary, our results indicate that CRP and SAA overexpression occurs in bronchial tissue and parenchyma in patients with COPD as compared to controls. Further, the expression differs in the parenchyma and bronchial tissue, and we provide evidence on the role of the vessels wall as a potential source of these biomarkers with a lack of correlation with the systemic inflammatory load. Future studies should address the cell types responsible for expressing these biomolecules, the mechanisms underlying this phenomenon, and its relevance in terms of systemic inflammatory load.
Abbreviations
COPD: Chronic obstructive pulmonary disease; CRP: C-reactive protein; FEV1: Forced expiratory volume in the first second; FVC: Forced vital capacity; RT-PCR: Reverse transcriptase-polymerase chain reaction; SAA: Serum amyloid A; SPSS: Statistical Package for Social Sciences
Acknowledgements
This study was financially supported by grants from the Fundación Neumosur (project number 03/2006) and the Consejería de Innovación, Ciencia y Empresa, Junta de Andalucía (Project number P08-CVI-3891).
Authors' contributions
JLLC is the main investigator and was responsible for project design, obtaining funding, completing and analyzing the database, and writing the manuscript. CC, BR, and DT selected the study patients and recorded the clinical information. MLP and AIB are thoracic surgeons who obtained the tissue samples. JS, VSL, AMW, and EA are biologists and were jointly responsible for carrying out the laboratory analysis.
Competing interests
None declared.
References
1. Nussbaumer-Ochsner Y, Rabe KF. Systemic manifestations of COPD. Chest. 2011;139:165-73 doi:10.1378/chest.10-1252
2. Garcia-Rio F, Miravitlles M, Soriano JB, Munoz L, Duran-Tauleria E, Sanchez G. et al. Systemic inflammation in chronic obstructive pulmonary disease: a population-based study. Respiratory research. 2010;11:63. doi:10.1186/1465-9921-11-63
3. Liu SF, Wang CC, Chin CH, Chen YC, Lin MC. High value of combined serum C-reactive protein and BODE score for mortality prediction in patients with stable COPD. Archivos de bronconeumologia. 2011;47:427-32 doi:10.1016/j.arbres.2011.04.011
4. Smith DJ, Yerkovich ST, Towers MA, Carroll ML, Thomas R, Upham JW. Reduced soluble receptor for advanced glycation end-products in COPD. The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 2011;37:516-22 doi:10.1183/09031936.00029310
5. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. The New England journal of medicine. 1999;340:448-54 doi:10.1056/NEJM199902113400607
6. Sinden NJ, Stockley RA. Systemic inflammation and comorbidity in COPD: a result of 'overspill' of inflammatory mediators from the lungs? Review of the evidence. Thorax. 2010;65:930-6 doi:10.1136/thx.2009.130260
7. Vernooy JH, Kucukaycan M, Jacobs JA, Chavannes NH, Buurman WA, Dentener MA. et al. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. American journal of respiratory and critical care medicine. 2002;166:1218-24
8. Urieli-Shoval S, Cohen P, Eisenberg S, Matzner Y. Widespread expression of serum amyloid A in histologically normal human tissues. Predominant localization to the epithelium. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 1998;46:1377-84
9. Paivaniemi OE, Maasilta PK, Vainikka TL, Alho HS, Karhunen PJ, Salminen US. Local C-reactive protein expression in obliterative lesions and the bronchial wall in posttransplant obliterative bronchiolitis. Mediators of inflammation. 2009;2009:510254. doi:10.1155/2009/510254
10. Ramage L, Proudfoot L, Guy K. Expression of C-reactive protein in human lung epithelial cells and upregulation by cytokines and carbon particles. Inhalation toxicology. 2004;16:607-13 doi:10.1080/08958370490464599
11. Bozinovski S, Uddin M, Vlahos R, Thompson M, McQualter JL, Merritt AS. et al. Serum amyloid A opposes lipoxin A(4) to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:935-40 doi:10.1073/pnas.1109382109
12. Leon-Atance P, Moreno-Mata N, Gonzalez-Aragoneses F, Canizares-Carretero MA, Garcia-Jimenez MD, Genoves-Crespo M. et al. Multicenter analysis of survival and prognostic factors in pathologic stage I non-small-cell lung cancer according to the new 2009 TNM classification. Archivos de bronconeumologia. 2011;47:441-6 doi:10.1016/j.arbres.2011.04.004
13. Malle E, De Beer FC. Human serum amyloid A (SAA) protein: a prominent acute-phase reactant for clinical practice. European journal of clinical investigation. 1996;26:427-35
14. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3:1101-8
15. Whitehead AS, de Beer MC, Steel DM, Rits M, Lelias JM, Lane WS. et al. Identification of novel members of the serum amyloid A protein superfamily as constitutive apolipoproteins of high density lipoprotein. The Journal of biological chemistry. 1992;267:3862-7
16. Kumon Y, Suehiro T, Hashimoto K, Sipe JD. Dexamethasone, but not IL-1 alone, upregulates acute-phase serum amyloid A gene expression and production by cultured human aortic smooth muscle cells. Scandinavian journal of immunology. 2001;53:7-12
17. Meek RL, Urieli-Shoval S, Benditt EP. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3186-90
18. Chen ES, Song Z, Willett MH, Heine S, Yung RC, Liu MC. et al. Serum amyloid A regulates granulomatous inflammation in sarcoidosis through Toll-like receptor-2. American journal of respiratory and critical care medicine. 2010;181:360-73 doi:10.1164/rccm.200905-0696OC
19. Dong Q, Wright JR. Expression of C-reactive protein by alveolar macrophages. Journal of immunology. 1996;156:4815-20
20. Venugopal SK, Devaraj S, Jialal I. Macrophage conditioned medium induces the expression of C-reactive protein in human aortic endothelial cells: potential for paracrine/autocrine effects. The American journal of pathology. 2005;166:1265-71 doi:10.1016/S0002-9440(10)62345-0
21. Lakota K, Resnik N, Mrak-Poljsak K, Sodin-Semrl S, Veranic P. Colocalization of serum amyloid a with microtubules in human coronary artery endothelial cells. Journal of biomedicine & biotechnology. 2011;2011:528276. doi:10.1155/2011/528276
22. Kim V, Han MK, Vance GB, Make BJ, Newell JD, Hokanson JE. et al. The chronic bronchitic phenotype of COPD: an analysis of the COPDGene Study. Chest. 2011;140:626-33 doi:10.1378/chest.10-2948
23. Bafadhel M, Umar I, Gupta S, Raj JV, Vara DD, Entwisle JJ. et al. The role of CT scanning in multidimensional phenotyping of COPD. Chest. 2011;140:634-42 doi:10.1378/chest.10-3007
24. McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG. et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. The New England journal of medicine. 2011;365:1567-75 doi:10.1056/NEJMoa1106955
25. Postma DS, Timens W. Remodeling in asthma and chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society. 2006;3:434-9 doi:10.1513/pats.200601-006AW
26. Han MK, Agusti A, Calverley PM, Celli BR, Criner G, Curtis JL. et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. American journal of respiratory and critical care medicine. 2010;182:598-604 doi:10.1164/rccm.200912-1843CC
27. Miravitlles M, Calle M, Soler-Cataluna JJ. Clinical phenotypes of COPD: identification, definition and implications for guidelines. Archivos de bronconeumologia. 2012;48:86-98 doi:10.1016/j.arbres.2011.10.007
28. Steel DM, Whitehead AS. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunology today. 1994;15:81-8 doi:10.1016/0167-5699(94)90138-4
29. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical chemistry. 2009;55:611-22 doi:10.1373/clinchem.2008.112797
30. Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Molecular and cellular biology. 1999;19:1720-30
31. Pinto-Plata VM, Mullerova H, Toso JF, Feudjo-Tepie M, Soriano JB, Vessey RS. et al. C-reactive protein in patients with COPD, control smokers and non-smokers. Thorax. 2006;61:23-8 doi:10.1136/thx.2005.042200
32. Wozniak K, Sleszycka J, Safianowska A, Wiechno W, Domagala-Kulawik J. Systemic inflammation in peripheral arterial disease with or without coexistent chronic obstructive pulmonary disease: analysis of selected markers. Archives of medical science: AMS. 2012;8:477-83 doi:10.5114/aoms.2012.29403
33. Young RP, Hopkins RJ, Whittington CF, Hay BA, Epton MJ, Gamble GD. Individual and cumulative effects of GWAS susceptibility loci in lung cancer: associations after sub-phenotyping for COPD. PloS one. 2011;6:e16476. doi:10.1371/journal.pone.0016476
34. Sundar IK, Mullapudi N, Yao H, Spivack SD, Rahman I. Lung cancer and its association with chronic obstructive pulmonary disease: update on nexus of epigenetics. Current opinion in pulmonary medicine. 2011;17:279-85 doi:10.1097/MCP.0b013e3283477533
35. de Torres JP, Marin JM, Casanova C, Cote C, Carrizo S, Cordoba-Lanus E. et al. Lung cancer in patients with chronic obstructive pulmonary disease-- incidence and predicting factors. American journal of respiratory and critical care medicine. 2011;184:913-9 doi:10.1164/rccm.201103-0430OC
Author contact
![]() Corresponding author: Jose Luis Lopez-Campos. Hospittal Universitario Virgen del Rocio, Avda. Manuel Siurot, s/n4013 Sevilla, Spain. Tel & Fax: 955013167 Email: lcamposes.
Corresponding author: Jose Luis Lopez-Campos. Hospittal Universitario Virgen del Rocio, Avda. Manuel Siurot, s/n4013 Sevilla, Spain. Tel & Fax: 955013167 Email: lcamposes.