Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(5):599-606. doi:10.7150/ijms.5151 This issue Cite
Research Paper
Propofol-Induced Protection of SH-SY5Y Cells against Hydrogen Peroxide Is Associated with the HO-1 via the ERK Pathway
Jing Gu, Meng Chi, Xuechao Sun, Guonian Wang ![]() , Mingming Li, Li Liu, Xuan Li
, Mingming Li, Li Liu, Xuan Li
Department of Anesthesiology, Third Affiliated Hospital of Harbin Medical University, No. 6 Baojian Rd., Nangang District, Harbin 150081, China.
Received 2012-9-3; Accepted 2013-3-7; Published 2013-3-19
Abstract
Propofol (2, 6-diisopropylphenol), is an anesthetic and routinely used for the humans sedation during surgery. The potent inducers of phase II detoxifying and antioxidant stress responsive to propofol were investigated. First, a dose of 25-100 µM propofol showed no significant cytotoxicity on SH-SY5Y cells and pre-treatment of SH-SY5Y cells with propofol (25-100 μM) for 8h prevented cell death and maintained cell integrity following exposure to 1 mM hydrogen peroxide by MTT assays. Then, an increase in the generation of ROS following hydrogen peroxide treatment was significantly attenuated by 8 h pre-treatment with propofol. Additionally, the potential roles of ERK, p 38 MAPK and JNK in the regulation of propofol-induced endogenous HO-1 expression in SH-SY5Y cells were estimated by Western blotting assays. Results showed that propofol significantly increased the phosphorylation levels of ERK, p 38 MAPK and JNK and antioxidant stress responsive to propofol was attenuated by the inhibition of ERK signaling biochemical inhibitors. These results suggest that the ERK pathway plays an important role in the regulation of propofol-mediated antioxidant effects in SH-SY5Y cells.
Keywords: Propofol, ERK, p 38 MAPK, JNK, SH-SY5Y cells.
1. Introduction
It is generally accepted that oxidative stress could induce cell damage and lead to a variety of neurodegenerative disorders such as Parkinson's and Alzheimer's diseases [1-3]. Hydrogen peroxide (H2O2) is one of the reactive oxygen species which could lead to lipid peroxidation and DNA damage [4, 5]. H2O2 could induce the production of reactive oxygen species (ROS) in the brain. Recent studies have demonstrated that high levels of ROS induce nervous system cell death [8-10]. Mammalian cells have developed several protective mechanisms involving both antioxidants and protective enzymes to defend ROS damage [11, 12]. HO-1 is one of antioxidant enzymes with potent anti-inflammatory, anti-oxidant and anti-proliferative effects [13-15]. HO-1 is known as a heat shock protein induced by the oxidative stress. In clinical practice, increased HO-1 expression prolongs cardiac xenograft survival, facilitates cold preservation of liver transplants and reduces the severity of ischemia reperfusion injury in kidney transplants [16-18]. In addition, overexpression of HO-1 also has effective neuroprotection against cold-induced secondary brain injury [19]. The expression of HO-1 could be induced by a range of substances, such as H2O2, phlorotannin compound, heavy metals, endotoxin, organic chemicals and so on [20-22]. Acquaviva et al. [23] showed that propofol induces expression of HO-1 in astrocytes under normal conditions. Expression of HO-1 has related to the several signal transduction pathways, such as mitogen-activated protein kinases (MAPKs) including extracellular signal-regulated kinases (ERKs), cJun-N-terminal kinases (JNKs) and p38-MAPK, depending on cell species and treatment conditions [24-27]. The expression also requires the activation of several signal transduction pathways such as protein kinase C (PKC) and phosphatidylinositol 3-kinase (PI3K) [28].
Propofol (2, 6-diisopropylphenol, Fig 1) was always described as an anesthetic [29]. Propofol produces a variety of pharmacodynamic effects, such as hypnosis to general anesthesia; relax amnestic and muscle; decrease the production of pro-inflammatory cytokines and alters the biosynthesis of nitric oxide (NO) [30, 31]. Additionally, propofol also could inhibit chemotaxis, attachment, migration, phagocytosis, the production of ROS and so on [32]. The protective effects of propofol were attributed to its capability of scavenging H2O2, reducing the formation of lipid peroxides, decreasing the expression levels of nitric oxide syntheses, and stabilizing the mitochondrial membrane [33, 34].
Structure of propofol.
In the present study, we investigated a mechanism by the protection of propofol for SH-SY5Y cells from H2O2 induced oxidative damage. Furthermore, the possible signaling way was examined to understand the mechanisms. We also investigated the effect of propofol on expression of HO-1 in SH-SY5Y cells under oxidative stress conditions.
2. Results and Discussion
2.1. Propofol inhibits H2O2-induced SH-SY5Y cell death
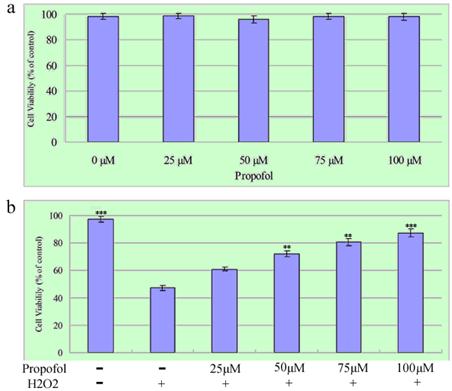
First, the cytotoxicity effects propofol on SH-SY5Y cells after 24 h exposure were examined (Figure 2a). The relative cell survival rate of SH-SY5Y cells treated with 0-100 µM propofol was over 80%, as analyzed by the MTT assay. Then, the effect of propofol on H2O2-induced cell death was determined. SH-SY5Y cells were treated with propofol (25-100 μM) or vehicle for 8 h, after that, the cells was exposed to 1 mM H2O2 or vehicle in fresh medium for 24 h. As shown in Fig. 2b, the survival rates of SH-SY5Y cells treated with 25 µM, 50 µM, 75 µM and 100 µM propofol were 60.94 %, 72.19 %, 80.43 % and 87.3 % respectively. The results indicate that a dose of 0-100 µM propofol does not contribute significantly to cytotoxicity in SH-SY5Y cells, and affords cytoprotection to the cell cultures in a concentration-dependent manner. Taken together, these data show that propofol inhibits H2O2-induced apoptosis in SH-SY5Y cells.
Propofol prevents H2O2-induced cell death; the results (presented as mean ± S.D.) are from three independent tests. (a) Cells were exposed to various concentrations of propofol for 24 h before being subjected to the MTT assay; (b) SH-SY5Y cells were treated with propofol (0-100 μM) for 8 h then incubated with H2O2 (1 mM) for a further 24 h, Cell viability was measured by MTT assays. ** and *** significant differences (P < 0.01 and P < 0.001 respectively) compared with 1 mM H2O2 group (the second bar).
2.2. Propofol Reduces H2O2-induced ROS Production of SH-SY5Y cell
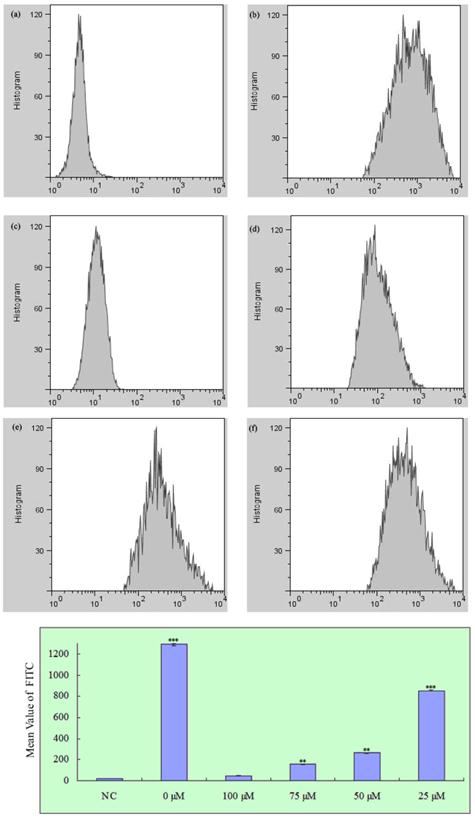
To determine whether propofol reduced H2O2-induced ROS generation in SH-SY5Y cells, cells were treated with propofol (0-100 μM) for 8 h and then incubated with 1 mM H2O2 or vehicle for 24 h. After that cells were treated with 25 μM H2-DCFDA for 1 h. As shown in Fig. 3, treatment with propofol significantly reduced the H2O2-induced ROS generation, indicating that propofol attenuates ROS production.
Effect of propofol on intracellular ROS formation in SH-SY5Y cells, fluorescence was detected by flow cytometry with FL1; results (presented as mean ± S.D.) are from three independent tests. ** and *** significant differences (P < 0.01 and P < 0.001 respectively) compared with NC group. (a) Negative control; (b)-(f) Treatment with 0, 100, 75, 50 and 25 µM propofol respectively.
2.3. ERK Signaling Pathways Participate in the propofol-Induced HO-1 Expression
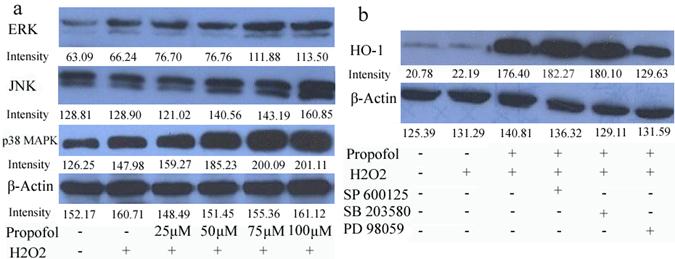
To analyze the signaling pathway that could participate in the neuroprotective mechanism of propofol, the effect of propofol on extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK) and p 38 mitogen-activated protein kinase (p 38 MAPK) activation in SH-SY5Y cells was investigated. Propofol stimulated concentration increases in ERK, JNK and p 38 MAPK phosphorylation in H2O2 induced SH-SY5Y cells (Fig 4). Moreover, PD98059 (an inhibitor of the ERK), SB203580 (an inhibitor of p 38 MAP kinase), and SP600125 (an inhibitor of JNK) were used in pretreatment of SH-SY5Y cells for 30 min, and then cells were co-treated with 100 µM propofol for another 8 h, cells were incubated for 24 h in the presence of H2O2 and cell lysates were resolved in SDS-PAGE and analyzed by immunoblot with anti-HO-1 antibody (Fig 5). Propofol increased the expression of HO-1, compared with control cells. Co-incubation of the cells with propofol, H2O2 and the ERK inhibitor PD98059 reduced the overexpression of HO-1, indicating that ERK was implicated in the overexpression of HO-1, induced by propofol. Moreover, JNK inhibitor and p 38 MAPK inhibitor did not block the overexpression of HO-1(Fig. 4-b). These results indicate that ERK is participating in the overexpression of HO-1 afforded by propofol.
ERK signaling pathway participates in the propofol-induced HO-1 expression; results (presented as mean ± S.D.) are from three independent tests. (a) Cells were treated with propofol of different concentrations. Total cell extracts were prepared and subjected to western-blot analysis in order to detect the active phosphorylated forms of p 38 MAPK, ERK1/2 and JNK; (b) Cells were treated with 10 µM SB203580, SP600125, or PD98059 for 30 min and challenged with 100 µM propofol for another 8 h then incubated with H2O2 for 24 h. Total cell extracts were prepared and subjected to western blot analysis for detection of the levels of HO-1.
ERK signaling pathway participate in the propofol reduces H2O2-induced SH-SY5Y cell death and ROS Production; results (presented as mean ± S.D.) are from three independent tests. (A) ERK signaling pathway participate in the propofol reduces H2O2-induced cell death. ** significant differences (P < 0.01) compared with 1 mM H2O2 group (the second bar). (B) The ROS production assay. (B-a) Blank control; (B-b) SH-SY5Y cell was treated with H2O2; (B-c) SH-SY5Y cell were treated with H2O2 and propofol; (B-d) The cells was treated with propofol for co-incubation, H2O2 and the ERK inhibitor PD98059. *** significant differences (P < 0.001) compared with the blank control group (the first bar).
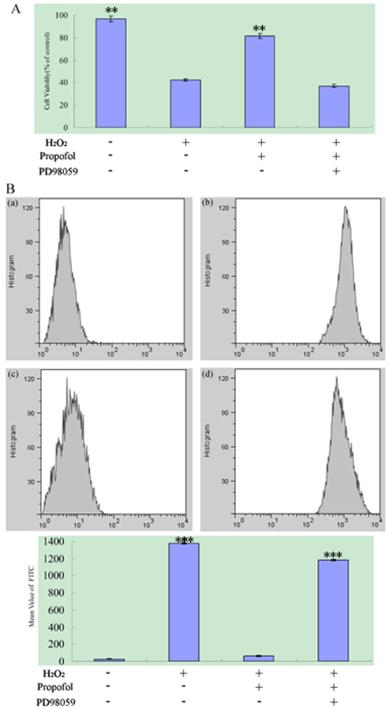
2.4 ERK Signaling Pathways Participate in the propofol Reduces H2O2-induced SH-SY5Y cell death and ROS Production
To confirm the role of ERK Signaling Pathways during oxidant-mediated apoptosis, we used H2O2 as the apoptosis inducer. According to our experiments described before, PD98059 (an inhibitor of the ERK) or vehicle were used in pretreatment of SH-SY5Y cells for 30 min, and then cells were co-treated with 100 µM propofol for another 8 h, after that, the cells was exposed to 1 mM H2O2 or vehicle in fresh medium for 24 h as analyzed by an MTT assay. For ROS production assay, PD98059 or vehicle were used in pretreatment of SH-SY5Y cells for 30 min, then cells were treated with 100 μM propofol or vehicle for 8 h and treated 1 mM H2O2 or vehicle for 24 h. Flowed that cells was treated with H2-DCFDA for 20 min, samples were analyzed by BD Flow cytometry with channel FL1, and the data were shown in Fig.5. From Fig.5, treatment with propofol significantly reduced the H2O2-induced SH-SY5Y cell death and ROS generation, but co-incubation of the cells with propofol, H2O2 and the ERK inhibitor PD98059 increase the SH-SY5Y cell death and ROS Production, indicating that the ERK inhibitor attenuates the effect of propofol in SH-SY5Y cell.
The purpose of this study was to elucidate the role of propofol in SH-SY5Y under oxidative stress. All data provide strong evidence that propofol could protect SH-SY5Y against oxidative stress via the ERK pathway and induction of the antioxidant enzyme HO-1. Propofol was able to protect neuroblastoma cells against oxidant stress as observed [35]. For example, the survival rates of SH-SY5Y cells treated with 25 µM-100 µM propofol were increase compared with cells untreated with propofol (47.11 %). This result refers that the high concentrations of propofol against H2O2 could be exerting a direct free radical scavenging effect. The antioxidant enzyme HO-1 was induced by propofol which related to the blocking ROS production as shown in the experiments with the fluorescent dye H2-DCFDA. Therefore propofol was able to reduce ROS production induced by H2O2 treatment both endogenous and exogenous [36]. HO-1 is an isoform which belongs to HO antioxidant enzyme induced in response to a variety of stress-inducing pathological conditions such as prolongs cardiac xenograft survival; facilitates cold preservation of liver transplants and so on. Maines [37] found that there is a low expression of HO-1 induced by H2O2 in neurons. Therefore, HO-1 represents a physiological protective mechanism against oxidative stress is generally accepted [38, 39]. In this study, propofol was able to up-regulate HO-1 and this effect was related to the protective effects of propofol.
The fact that propofol-induced protective effect was prevented by the ERK inhibitor indicates that ERK pathway is participating in the induction of antioxidant enzyme HO-1. Our results are consistent with those which were found for the propofol protected cells against ischemia/reperfusion injury in liver epithelial cells [40]. Based on our present results, propofol exerts a protective effect by modulating signaling pathways in neuronal cells under oxidative stress. Collectively, the data suggest that propofol reduced cell death and ROS production which induced by H2O2 via ERK pathway. Further studies are needed to delineate the detailed mechanisms of activating ERK pathway by propofol, and which protein was in cooperation with HO-1 during oxidative stress-induced neuronal to reduce cell death.
In summary, this study shows that propofol can afford protection of SH-SY5Y neuroblastoma cells under oxidative stress conditions by induction of the antioxidant enzyme HO-1 via ERK pathway. This finding may have neuroprotective properties that might be useful in the acute treatment of stroke, to prevent neuronal damage and the ensuing neurological sequelae.
3. Experimental
3.1. Cell Culture
The human neuroblastoma SH-SY5Y cells were maintained in MEM medium supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 U/ml streptomycin in a humid atmosphere of 5% CO2 and 95% air at 37 °C. SH-SY5Y cells were plated in plates (10 cm). Then cells were pretreated with various concentrations (0-100 μM) of propofol for 8 h, followed by exposure to 1 mM of H2O2 in the presence of the same concentrations of propofol for another 24 h. To produce oxidative stress, H2O2 was freshly prepared from 30% stock solution prior to each experiment. The control cells were added with the same medium without H2O2 and propofol.
3.2. Chemicals and Antibodies
Propofol was prepared from Diprivan (Zeneca Limited, Macclesfield, and Cheshire, UK). The vehicle contained glycerol, soybean oil, purified egg phosphatide/egg lecithin, sodium hydroxide, and water. 3-[4, 5-dimethyl-2-thiazolyl]-2, 5-diphenyl-2-tetrazolium bromide (MTT) were purchased from Sigma. Other drugs and reagents used in this study were as follows: PD98059 (an inhibitor of the ERK), SB203580 (an inhibitor of p 38 MAP kinase), and SP600125 (an inhibitor of JNK) [41] were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against HO-1 and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal beta-actin antibody was obtained from Chemicon (Temecula, CA, USA). All other reagents were from commercial suppliers and of standard biochemical quality.
3.3. Viability Assay
SH-SY5Y cells were plated at a density of 1×104 cells per well in 96-well plates, and the cell viability was determined using the conventional MTT reduction assay. Briefly, after 24 h exposure to H2O2, 40 μl of MTT (2 mg/ml in PBS) were added to each well and the cells were incubated at 37 °C for 4 h. The supernatants were aspirated carefully and 100 μl of dimethyl sulfoxide (DMSO) were added to each well to dissolve the precipitate and the absorbance at 490 nm was measured with a microplate reader (BIO-RAD Model 3550, CA, USA). The cell viability ratio was calculated by the following formula: Inhibitory ratio (%) = [(ODcontrol − ODtreated) / (ODcontrol)] × 100%.
3.4. Flow cytometric detection of reactive oxygen species (ROS)
ROS production in SH-SY5Y cells was measured using the redoxsensitive fluorescent dye H2- DCFDA. After pretreatment with propofol or vehicle 8 h, then treatment with 1 mM H2O2 for 24 h, cells were incubated with 25 μM H2-DCFDA for 20 min. The cells were rinsed twice with phenol-red-free DMEM containing 1% FBS, and fluorescence was detected on flow cytometry with FL1.
3.5. Western-Blot Analysis
For isolation of protein fractions, SH-SY5Y cells (1 × 106) were seeded in a 6 cm plate and incubated at 37 °C for 24 h. Cells were pretreated with various concentrations of propofol for 8 h and then washed twice with ice-cold PBS, and lysed using cell lysis buffer [20 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM EDTA, 1% Na3CO4, 0.5 g/mL leupeptin, 1 mM phenylmethanesulfonyl fluoride (PMSF)]. The lysates were collected by scraping from the plates and then centrifuged at 10,000 rpm at 4 °C for 5 min. The protein samples (20 μg) were loaded on a 12% of SDS-polyacrylamide gel for electrophoresis, and transferred onto PVDF transfer membranes (Millipore, Billerica, MA, USA) at 0.8 mA/cm2 for 2 h. Membranes were blocked at room temperature for 2 h with blocking solution (1% BSA in PBS plus 0.05% Tween-20). Membranes were incubated overnight at 4 °C with primary anti-bodies at a 1:1000 dilution (Biosynthesis Biotechnology Company, Beijing, China) in blocking solution. After three washings in TBST for each 5 min, membranes were incubated for 1 h at room temperature with an alkaline phosphatase peroxidase-conjugated anti-mouse secondary antibody at a dilution of 1:500 in blocking solution. Detection was performed by the BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime Institute of Biotechnology, Suzhou, China) according to the manufacturer's instructions. Bands were recorded by a digital camera (Canon, EOS 350D, Tokyo, Japan).
3.6. Statistical Analyses
Data are presented as means ± SEM. Differences were evaluated using unpaired Student's t-tests. The level of statistical significance was set at p < 0.05.
Abbreviation
HO-1: heme oxygenase-1; PKC: protein kinase C; H2O2: hydrogen peroxide; ROS: reactive oxygen species.
Acknowledgements
The authors gratefully acknowledge the financial supports by the topic of Heilongjiang Provincial Education Bureau (Jing.Gu), the Research projects of Heilongjiang Provincial Health Bureau (2013-649).
Conflict of Interest
The authors declare no conflict of interest.
References
1. Keum YS, Owuor ED, Kim BR, Hu R, Kong AN. Involvement of Nrf2 and JNK1 in the activation of antioxidant responsive element (ARE) by chemopreventive agent phenethyl isothiocyanate (PEITC). Pharm Res. 2003;20:1351-1356
2. Kong AN, Owuor E, Yu R. Induction of xenobiotic enzymes by the MAP kinase pathway and the antioxidant or electrophile response element (ARE/EpRE). Drug Metab Rev. 2001;33:255-271
3. Hu R, Hebbar V, Kim BR. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther. 2004;310:263-271
4. Ahmed K, Gerber DA, Cochet C. Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol. 2002;12:226-230
5. Alia M, Ramos S, Mateos R, Bravo L, Goya L. Response of the antioxidant defense system to tert-butyl hydroperoxide and hydrogen peroxide in a human hepatoma cell line (HepG2). J. Biochem Mol Toxicol. 2005;19:119-128
6. Riveiro ME, De Kimpe N, Moglioni A, Vazquez R, Monczor F, Shayo C, Davio C. Coumarins: old compounds with novel promising therapeutic perspectives. Curr Med Chem. 2010;17:1325-1328
7. Satoh T, Enokido Y, Aoshima H, Uchiyama Y, Hatanaka H. Changes in mitochondrial membrane potential during oxidative stressinduced apoptosis in PC12 cells. J Neurosci Res. 1997;50:413-420
8. Bourbon NA, Sandirasegarane L, Kester M. Ceramide-induced inhibition of Akt mediated through protein kinase C. J Biol Chem. 2002;277:3286-3292
9. Uberti D, Piccioni L, Colzi A, Bravi D, Canonico PL, Memo M. Pergolide protects SH-SY5Y cells against neurodegeneration induced by H2O2. Eur J Pharmacol. 2002;434:17-20
10. Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2-14
11. Doornbos RP, Theelen M, Van der Hoeven PC, Van Blitterswijk WJ, Verkleij AJ, van Bergen en Henegouwen PMP. Protein kinase C is a negative regulator of protein kinase B activity. J Biol Chem. 1999;2274:8589-8596
12. Ha JH, Noh HS; Shin IW, Hahm JR, Kim DR. Mitigation of H2O2-induced autophagic cell death by propofol in H9c2 cardiomyocytes. Cell biology and toxicology. 2012;28(1):19-29
13. Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP 600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci. 2001;98:13681-13686
14. Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699-22704
15. Strasser A, O'Connor L, Dixit VM. Apoptosis signalling. Annu Rev Biochem. 2000;69:217-245
16. Casamayor A, Morrice NA, Alessi DR. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem J. 1999;342:287-292
17. Encinas M, Iglesias M, Llecha N, Comella JX. Extracellular regulated kinase and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neutritogenesis of the neuroblastoma cell line SH-SY5Y. J Neurochem. 1999;73:1409-1421
18. Eckenhoff RG, Johansson JS, Wei H, Carnini A, Kang B, Wei W, Pidikiti R, Keller JM, Eckenhoff MF. Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology. 2004;101:703-709
19. Crossthwaite AJ, Hasan S, Williams RJ. Hydrogen peroxide-mediated phosphorylation of ERK1/2, Akt/PKB and JNK in cortical neurones: dependence on Ca2+ and PI3-kinase. J Neurochem. 2002;80:24-35
20. Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci. USA. 2003;100:721-726
21. Paya M, Halliwell B, Hoult JR. Interactions of a series of coumarins with reactive oxygen species. Scavenging of superoxide, hypochlorous acid and hydroxyl radicals. Biochem Pharmacol. 1992;44:205-214
22. Wang X, Zhang X, Cheng Y, Li C, Zhang W, Liu L, Ding Z. Alpha-lipoic acid prevents bupivacaine-induced neuron injury in vitro through a PI3K/Akt-dependent mechanism. Neurotoxicology. 2010;31(1):101-112
23. Acquaviva R, Campisi A, Murabito P, Raciti G, Avola R, Mangiameli S, Musumeci I, Barcellona ML, Vanella A, Li VG. Propofol attenuates peroxynitrite-mediated DNA damage and apoptosis in cultured astrocytes: an alternative protective mechanism. Anesthesiology. 2004;101:1363-1371
24. Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? Journal of Signal Transduction. 2011 doi:10.1155/2011/792639
25. Cowan KJ, Storey KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. The Journal of Experimental Biology. 2003;206:1107-1115
26. Chang AYW. Pro-life role for c-Jun N-terminal kinase and p38 mitogen-activated protein kinase at rostral ventrolateral medulla in experimental brain stem death. Chang Journal of Biomedical Science. 2012;19:1-12
27. Wu XJ, Zheng YJ, Cui YY, Zhu L, Lu Y, Chen HZ. Propofol attenuates oxidative stress-induced PC12 cell injury via p38 MAP kinase dependent pathway. Acta pharmacologica Sinica. 2007;28(8):1123-1128
28. Cakatay U. Pro-oxidant actions of alpha-lipoic acid and dihydrolipoic acid. Med Hypotheses. 2006;66(1):110-117
29. Tamagno E, Robino G, Obbili A, Bardin P, Aragno M, Parola M, Danni O. H2O2 and 4-hydroxynonenal mediate amyloid beta-induced neuronal apoptosis by activating JNKs and p38 MAPK. Exp Neurol. 2003;180:144-155
30. Uberti D, Rizzini C, Spano PF, Memo M. Characterization of tau proteins in human neuroblastoma SH-SY5Y cell line. Neurosci Lett. 1997;235:149-153
31. Brunet A, Datta SR, Greenberg ME. Transcription-dependent and independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297-305
32. Magrané J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24:1700-1706
33. Do SH, Ham BM, Zuo Z. Effects of propofol on the activity of rat glutamate transporter type 3 expressed in Xenopus oocytes: the role of protein kinase C. Neurosci Lett. 2003;343:113-116
34. Chikutei K, Oyama TM, Ishida S, Okano Y, Kobayashi M, Matsui H, Horimoto K, Nishimura Y, Ueno SY, Oyama Y. Propofol, an anesthetic possessing neuroprotective action against oxidative stress, promotes the process of cell death induced by H2O2 in rat thymocytes. European journal of pharmacology. 2006;540(1-3):18-23
35. Dudek H, Datta SR, Franke TF, Birnbaum MJ, Ryoji Y, Cooper GM, Segal RA, kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661-665
36. Ishii T, Yanagawa T, Tetsuya K, Yuki K, Seita J, Yoshida H, Bannai S. Murine peritoneal macrophages induce a novel 60-kDa protein with structural similarity of a tyrosine kinase p56 lck -associated protein to oxidative stress. Biochem Biophys Res Commun. 1996;226:456-460
37. Maines M. The heme oxygenase system: a regulator of second messenger gases. Ann Rev Pharmacol Toxicol. 1997;37:517-554
38. Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates and functions during apoptosis. Annu Rev Biochem. 1999;68:383-424
39. Wang HX, Zhang G, Wang Q, Feng XS, Zong H. Propofol protects hepatic L02 cells from hydrogen peroxide-induced apoptosis via activation of extracellular signal-regulated kinases pathway. Anesthesia and analgesia. 2008;107(2):534-40
40. Xu JJJ, Wang YLL. Propofol attenuation of hydrogen peroxide-mediated oxidative stress and apoptosis in cultured cardiomyocytes involves haeme oxygenase-1. European journal of anaesthesiology. 2008;25(5):395-402
41. Liang C, Xue ZHG, Wang H, Li P. Propofol Upregulates Heme Oxygenase-1 Through Activation of ERKs in Human Umbilical Vein Endothelial Cells Under Oxidative Stress Conditions. Laboratory Investigation. 2011;23(3):229-235
Author contact
![]() Corresponding author: Dr. Guonian Wang, Department of Anesthesiology, Third Affiliated Hospital of Harbin Medical University, No. 6 Baojian Rd., Nangang District, Harbin 150081, China. wangguonian609cncom.cn; Tel.: +86-0451-86298332.
Corresponding author: Dr. Guonian Wang, Department of Anesthesiology, Third Affiliated Hospital of Harbin Medical University, No. 6 Baojian Rd., Nangang District, Harbin 150081, China. wangguonian609cncom.cn; Tel.: +86-0451-86298332.