Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Why and how do we measure HbA1c?
Can HbA1c be used for diagnosing...
Which factors other than...
What are the consequences of...
Does chronic hyperglycemia in...
Is lowering of HbA1c the key to...
Reduction of glycemic...
Conclusion
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2012; 9(8):665-681. doi:10.7150/ijms.4520 This issue Cite
Review
What Do We Need beyond Hemoglobin A1c to Get the Complete Picture of Glycemia in People with Diabetes?
Rolf Hinzmann* ![]() , Christof Schlaeger*, Cam Tuan Tran*
, Christof Schlaeger*, Cam Tuan Tran*
Roche Diagnostics GmbH, Mannheim, Germany.
* All authors contributed equally to this work and are employees at Roche Diagnostics GmbH.
Received 2012-4-24; Accepted 2012-8-19; Published 2012-9-29
Abstract
Hemoglobin A1c (HbA1c) is currently the most commonly used marker for the determination of the glycemic status in people with diabetes and it is frequently used to guide therapy and especially medical treatment of people with diabetes. The measurement of HbA1c has reached a high level of analytical quality and, therefore, this biomarker is currently also suggested to be used for the diagnosis of diabetes. Nevertheless, it is crucial for people with diabetes and their treating physicians to be aware of possible interferences during its measurement as well as physiological or pathological factors that contribute to the HbA1c concentration without being related to glycemia, which are discussed in this review. We performed a comprehensive review of the literature based on PubMed searches on HbA1c in the treatment and diagnosis of diabetes including its most relevant limitations, glycemic variability and self-monitoring of blood glucose (SMBG). Although the high analytical quality of the HbA1c test is widely acknowledged, the clinical relevance of this marker regarding risk reduction of cardiovascular morbidity and mortality is still under debate. In this respect, we argue that glycemic variability as a further risk factor should deserve more attention in the treatment of diabetes.
Keywords: Hemoglobin A1C, diabetes mellitus, hemoglobinopathies, glycemic control, glycemic variability, diabetes complications.
Why and how do we measure HbA1c?
Hemoglobin A1c (HbA1c, or A1c) is currently the most prominent biomarker for assessing the glycemic status of people with diabetes and for making decisions on the appropriate therapy adjustments, if needed. Discovered more than forty years ago by Samuel Rahbar [1] and co-workers, the breakthrough for HbA1c was achieved when it was discovered in the Diabetes Control and Complications Trial (DCCT) in 1993 that the concentration of HbA1c was an excellent predictor of diabetes-related long-term complications [2]. Over the years, many healthcare providers have come to view the HbA1c value as a "magic number" that comprises all of the information required for managing blood glucose concentrations to prevent complications in people with diabetes; the concept "the lower the better" was considered a tempting approach.
With this review article we do not intend to question the value of HbA1c measurements; rather it is to discuss some limitations of this biomarker that healthcare providers should be aware of. These limitations are related to the analytical measurement of HbA1c and to a variety of physiological and pathological conditions influencing the HbA1c concentration.
Glucose exists predominantly in a cyclic form; however, this form is in chemical equilibrium with a small fraction of acyclic glucose which exhibits a very chemically active aldehyde group that is able to react in a non-enzymatic manner with amino groups of the side chains of proteins. The reaction takes place in two steps (Fig. 1): In the first, reversible step, glucose forms an aldimine (Schiff base) with an amino group in the protein. In the second, irreversible step, the aldimine becomes a ketoamine which persists for the remaining lifetime of the protein. This chemical reaction is termed glycation.
The formation of glycated hemoglobin is a non-enzymatic reaction consisting of two steps of which only the first one is reversible.
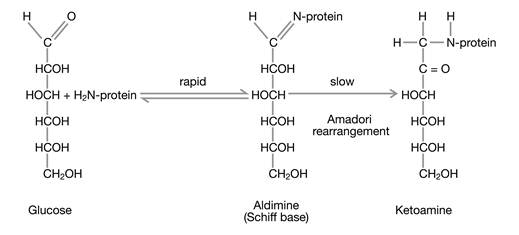
In principal, all amino groups of the side chains of proteins can become glycated; however, a couple of factors have an important impact on the concentrations of the final glycation end products: (a) the concentration of the protein, (b) the steric accessibility of the different side chain amino groups, (c) the glucose concentration in the compartment of the reaction, and (d) the lifespan of the glycated protein.
Hemoglobin makes up most of the content of red blood cells, which are continuously exposed to glucose in the blood. Therefore, hemoglobin is also continuously becoming glycated. What Samuel Rahbar and co-workers had seen was a thin extra band on electrophoresis gels, representing glycated hemoglobin. Initially, HbA1c was just the name given to a band in hemoglobin electrophoresis, and only later it was found that it consists of a mixture of hemoglobin molecules that are glycated at different side chain amino positions. Assuming that the reaction rate is proportional to the hemoglobin concentration and that the accessibility of the side chain amino groups of hemoglobin for glucose and the lifetime of the red blood cells are constant, only the glucose concentration should have an influence on the concentration of HbA1c in terms of percentage. Accordingly, HbA1c would be the perfect proxy for the blood glucose concentration over the average lifespan of red blood cells. However, we now know that this model is an oversimplified description.
As the quantification of the small HbA1c peak in comparison to the large HbA0 peak is difficult to perform in gel electrophoresis, more advanced technologies have been developed. Today, most HbA1c measurements are performed using either ion exchange high performance liquid chromatography (HPLC) in which HbA1c is separated from HbA0 due to its different electric charge, by boronate affinity column methods where the glycosylated hemoglobins are bound by immobilized boronic acid to separate them from non-glycosylated hemoglobins, or by immunoassays using antibodies against glycated hemoglobin and turbidimetry, for example, as the detection principle. It is important to note that certain routine laboratory methods may give inaccurate results in the presence of pathological hemoglobins such as HbC, HbS, HbE or HbD (see below).
When HbA1c became a global standard for determining glycemic status after the DCCT was published, it became apparent from external quality assessment schemes that HbA1c concentrations determined from identical blood samples in external quality assessment schemes showed (sometimes striking) differences in different laboratories, thus limiting the diagnostic value of HbA1c determination.
To overcome this obstacle, two different approaches were adopted (historical reviews [3,4]). In 1996, the National Glycohemoglobin Standardization Program (NGSP) [5,6] was launched in the USA. The NGSP consists of primary and secondary reference laboratories running a special HPLC method as the designated comparison method. Manufacturers and clinical laboratories that intend to reach NGSP certification must fulfill certain precision requirements and must recover samples within pre-defined limits in method comparisons of sample panels distributed by NGSP reference laboratories. The allowed deviations have become smaller over the years.
In 1994, the International Federation for Clinical Chemistry and Laboratory Medicine (IFCC) adopted an entirely different approach [7]. Because HbA1c is a combination of hemoglobins glycated at different side chain amino groups, which makes it difficult to standardize the analyte, an IFCC working group redefined HbA1c in the following way: HbA1c is HbA0 glycated at the N-terminal amino group of the β chain (βN-1-deoxyfructosyl-hemoglobin).
The advantage of this definition is that HbA1c is now no longer considered a mixture but a clearly identified chemical substance, comprising more than 60% of the amount of substance that has previously been termed HbA1c. The disadvantage is that the numerical value of the concentration has changed and is, therefore, not comparable to what it had been before and that the concentrations measured by a newly developed reference procedure (see next paragraph) are no longer comparable to those obtained during the DCCT. However, this issue has been addressed and solved.
Once the analyte HbA1c had been redefined, another working group within the IFCC developed a reference system for HbA1c, comprising: (a) a primary reference material [8], (b) a reference method ("reference procedure") [9], and (c) a network of reference laboratories [10]. In short, the reference procedure is based on the specific enzymatic cleavage of the glycated and non-glycated hemoglobin, resulting in both a glycated and a non-glycated β-N-terminal hexapeptide. These hexapeptides are first separated from the remaining peptides in the sample by reversed phase HPLC and then quantified during either capillary electrophoresis or electron spray ionization mass spectrometry. Only few laboratories, globally, are capable of performing this reference procedure.
In a consensus statement [11], the IFCC, the International Diabetes Federation (IDF), the International Society for Pediatric and Adolescent Diabetes (ISPAD), the American Diabetes Association (ADA), and the European Association for the Study of Diabetes (EASD) stated that this reference procedure is the "metrological anchor" for HbA1c measurement, and that measurement results shall be expressed in mmol/mol and in addition in percent. In order to avoid a change of the numerical values, the HbA1c percent values are now recalculated to match former “DCCT values” using the equation HbA1c (%) = [0.915*IFCC reference method value (%)] + 2.15 so that the change in standardization does not change the numeric HbA1c concentrations that patients and healthcare providers are already familiar with. As the IFCC reference procedure has now been accepted as the metrological anchor for the different national HbA1c standardization schemes, including NGSP, HbA1c is now recognized as a protein with a very high level of standardization compared to other proteins used in clinical laboratory medicine.
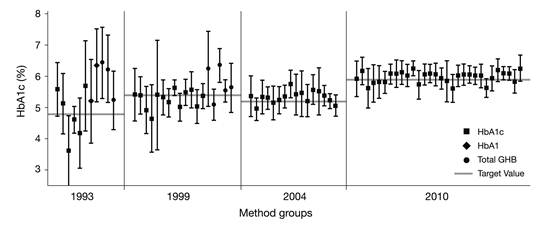
The result of this standardization can be seen immediately when recent results of external quality assessment schemes are compared to earlier ones (e.g., as seen in laboratory performance data from surveys of the College of American Pathologists [CAP] [12] from 1993, 1999, 2004, and 2010) (Fig. 2). The inter- laboratory standard deviation has significantly improved. The quality of HbA1c measurement is generally considered satisfying, except for some point-of-care methods in the market [13]. This was one of the prerequisites for the suggestion to use HbA1c not only as marker for monitoring the metabolic status of people with diabetes but also for the diagnosis of diabetes.
Inter-laboratory standard deviations in external quality assessment schemes (here surveys of the American College of Pathologists [CAP]) [12] have decreased over time due to implementation of standardization and decreasing random error of HbA1c measurement devices.
Can HbA1c be used for diagnosing diabetes?
The National Diabetes Data Group (NDDG) provided consensus diagnostic criteria for diabetes in 1979 that were based on distributions of glucose levels, rather than on the relationship of glucose levels with complications. The diagnostic cut-off values chosen were based on the relative risk of decompensation to overt or symptomatic diabetes. These criteria comprised a fasting plasma glucose (FPG) of 140 mg/dL (7.8 mmol/L) or greater and a 2-h plasma glucose (PG) concentration during a 75 g oral glucose tolerance test (OGTT) of 200 mg/dL (11.1 mmol/L) or greater [14]. Later, in 1997, the Expert Committee on Diagnosis and Classification of Diabetes Mellitus reviewed existing population data for retinopathy in three different populations - Egyptians, Pima Indians and the U.S. National Health and Nutrition Examination Survey (NHANES) population - and determined that the relatively diabetes-specific comorbidity retinopathy was not detected at lower levels of FPG, 2-h PG, and HbA1c concentrations; above certain thresholds, retinopathy prevalence increased progressively. Accordingly, the group recommended an FPG of 126 mg/dL (7.0 mmol/L) and confirmed the existing 2-h PG threshold of 200 mg/dL to be adequate cut-off values for the diagnosis of diabetes [15]. In the following decade, those threshold concentrations became worldwide standards for the diagnosis of diabetes. The use of HbA1c was not recommended for the diagnosis of diabetes due to a lack of worldwide standardization of the laboratory assay and absence of consensus on the appropriate diagnostic cut-off value.
More than a decade later, in 2009, the International Expert Committee with members appointed by the ADA, the EASD, and the IDF convened and recommended an HbA1c cut-off value of 6.5% (48 mmol/mol) for the diagnosis of diabetes [16]. This decision was based on an analysis of the DETECT-2 Study, a database analysis of 13 studies examining the association of retinopathy with measures of glycemia, assessed and graded by fundus photography and comprising data from 28,898 persons aged 20 to 79 [16].
In its 2010 Clinical Practice Recommendations, the ADA included an HbA1c of 6.5% (48 mmol/mol) or greater as a diagnostic criterion for diabetes. In addition to the availability of comprehensive population data on retinopathy prevalence in association with HbA1c concentrations, these recommendations were enabled due to significant proceedings in the attempt to establish worldwide standardization of the HbA1c assay. Furthermore, with advances in instrumentation as well as accuracy and precision, the performance of the current state of the art HbA1c assays match those of glucose assays. In addition, HbA1c testing offers many advantages over FPG or 2-h PG tests for the diagnosis of diabetes [17]:
- Standardization and alignment with the DCCT / United Kingdom Prospective Diabetes Study (UKPDS);
- Better index of overall glycemic exposure;
- Equal or greater accuracy in predicting risk for long-term complications (retinopathy);
- Significantly less biologic variability;
- Lower level of short-term intra-individual variability [18];
- High pre-analytical stability (up to one week at 4°C);
- Ability to sample blood at any time — fasting samples are no longer required;
- Relatively unaffected by acute fluctuations in glucose levels (e.g., due to stress or dietary intake);
- Established for guiding diabetes management and decision support;
- Considerably quicker to obtain than OGTT results.
There is broad consensus on the advantages of a standardized HbA1c assay over FPG and 2-h PG testing, however, the most appropriate cut-off value for the diagnosis of diabetes is still subject to debate. The International Expert Committee based their decision to establish the diagnostic cut-off value as 6.5% (48 mmol/mol) on an analysis of the DETECT-2 Study [16] and on three population analyses that were included in the 1997 report. When the data for moderate non-proliferative diabetic retinopathy (NPDR) were examined in 0.5% increments, the glycemic level at which the prevalence began to rise above background levels turned out to be 6.5% (48 mmol/mol) [17]. Moderate retinopathy was practically nonexistent in more than 20,000 subjects with HbA1c concentrations <6.5%. Conclusively, the optimal cut-off value for detecting at least moderate retinopathy was an HbA1c of 6.5%. According to the International Expert Committee, this threshold provides a fair balance for the potential stigma and costs of mistakenly identifying individuals as people with diabetes and the clinical consequences of delaying the diagnosis in someone with an HbA1c concentration <6.5% (48 mmol/mol).
There is no doubt that HbA1c concentrations between subjects with normal OGTT and those with diabetes or impaired glucose tolerance (IGT) show some overlap, as demonstrated by Fajans et al. [19]. According to their data analysis, HbA1c may be in the normal range in subjects with diabetes mellitus or IGT and among subjects with mild fasting hyperglycemia. Therefore, they claim that HbA1c alone is not a sufficiently reliable tool for recognizing particularly the early stages of diabetes or IGT and recommend that plasma glucose determinations (FPG, 2-h PG, or OGTT) should be used if a history or symptoms indicate a high risk for the presence of diabetes and HbA1c is <6.5% (48 mmol/mol).
Other studies examining the utility of the HbA1c assay within different ethnic groups, including Chinese, Arabs, and non-Hispanic white and black Americans, suggest that a universal diagnostic cut-off value of 6.5% (48 mmol/mol) may not be appropriate to identify all people at risk of developing retinopathy who should, therefore, be diagnosed as having diabetes [19-21]. Based on the NHANES-3 data from 2005 to 2006, it was estimated that 5.9 million non-Hispanic U.S. adults with unrecognized diabetes would be missed by screening with the HbA1c method and the proposed diagnostic cut-off values only [21].
Conclusively, it should be pointed out that HbA1c is a relatively weak surrogate marker for glucose but rather serves as an alternative marker for microvascular risk [22]. In our view, this is supported by the findings of the A1c-Derived Average Glucose (ADAG) Study Group. When describing the concept of 'Translating the A1c Assay Into Estimated Average Glucose Values' [23], it became apparent that although 90% of HbA1c concentrations predicted the average measured glucose within ± 15.0%, significant deviations were observed (Fig. 3). Since the average glucose concentration had been determined very carefully in this study, these deviations are hard to explain by measurement error only.
The A1c-Derived Average Glucose (ADAG) Study Group demonstrated that HbA1c correlates well with average glucose (AG) (R2 = 0.84) [23], however, although 90% of HbA1c concentrations predicted the average measured glucose within ± 15%, significant deviations were observed. The regression equation is: Calculated AG (mg/dL) = 28.7 x HbA1c (%) - 46.7.
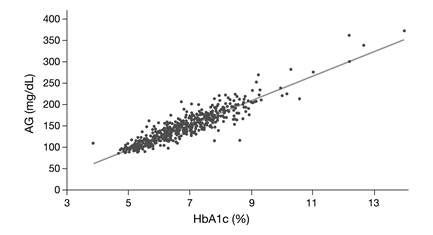
As a consequence, HbA1c detects different populations at risk, which is not essentially a concern, given that it predicts risk equally well. However, due to ethnic differences in the relationship between HbA1c and mean blood glucose [24], a universally defined threshold for the diagnosis of diabetes will remain subject of debate.
Which factors other than glycemia can affect HbA1c concentrations?
In people with diabetes with an HbA1c >7.0% (53 mmol/mol) on oral anti- diabetic therapy, basal hyperglycemia contributes most to HbA1c; after addition of basal insulins, basal hyperglycemia still contributes one third to HbA1c [25]. However, despite all progress in analytical standardization of HbA1c, the limitation still remains that the concentration of HbA1c is affected by other factors than glucose.
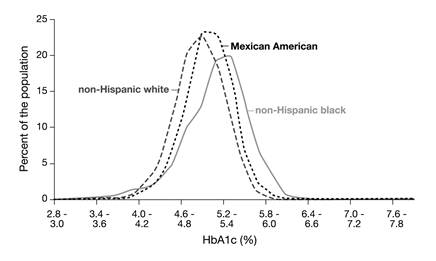
Before glycation of hemoglobin can take place, glucose has to move across the cell membrane into the interior of the red blood cells. This is a fast process mediated by the passive glucose transporter GLUT1 [26]. However, people with identical blood glucose concentrations may have different glucose concentrations in their red blood cells [27]. If the kinetic characteristics of glucose transport across the red blood cell were equal for all individuals, the ratio between the concentrations of (intracellular) HbA1c and (extracellular) glycated serum albumin (also termed fructosamine) should be an intra- and inter-individual constant. It has, however, been shown that this ratio remains constant for individuals only but not for populations; an observation that has also been called the 'glycation gap' [28]. This factor may, among others, also explain the differences that have been observed in the distribution of HbA1c concentrations in non-diabetic non-Hispanic white, non-Hispanic black and Mexican Americans in the NHANES-3, that cannot be explained by other confounding factors (Fig. 4) [29,30].
The distributions of HbA1c concentrations in non-diabetic non-Hispanic white, non-Hispanic black and Mexican Americans in the National Health and Nutrition Examination Survey-3 (NHANES-3) were different, with non-Hispanic blacks having the highest values [29].
Finally, twin studies have contributed to our understanding that there is an inherited component contributing to individual HbA1c concentrations. The correlation of HbA1c concentrations in non-diabetic monozygotic twins was higher than in non-diabetic dizygotic twins (r = 0.77 vs. r = 0.53), and HbA1c was not only correlated in monozygotic twins concordant for type 1 diabetes (T1DM) (r = 0.68) but, to a smaller degree, also in monozygotic twins discordant for T1DM (r = 0.52) [31]. The authors concluded that genetic effects (heritability) account for 62% of the variance of HbA1c in the study population; the rest is attributable to environment and age. Surprisingly, HbA1c heritability could not be attributed to genes related to fasting glucose.
As red blood cells typically have a lifespan of about 120 days, HbA1c should represent glycemia over this period of time. Obviously, all conditions that shorten the lifespan of red blood cells will decrease HbA1c as the average time during which red blood cells are exposed to glucose is reduced. Such conditions comprise, for example, hypersplenism and all types of hemolytic anemias which may be acquired (e.g., during infections such as malaria, in hemolytic microangiopathies, in drug- induced or in auto-immune hemolytic anemia), or that are inborn (e.g., variants of the hemoglobin genotype in hemoglobinopathies, disturbed hemoglobin synthesis in thalassemias, or increased red blood cell fragility in elliptocytosis and spherocytosis). There are indications that hyperglycemia itself may also reduce red blood cell lifespan [32].
According to the World Health Organization (WHO), at least 5.2% of the world population carries a significant hemoglobin variant [33]. The hemoglobin variants comprise a variety of congenital disorders of one or more of the four globin chains of hemoglobin. In general, hemoglobinopathies are divided into two groups: thalassemia syndromes and abnormal hemoglobins. Both are caused by mutations and/or deletions in the α- or β-globin genes, leading to a reduced rate of synthesis or no synthesis of one of the globin chains and/or differences in the amino acid composition, while the hem group remains unchanged. When gene defects cause hemoglobin synthesis disorders, this gives rise to thalassemias. The hemoglobin structure in these cases is normal [34]. When mutations cause changes in the hemoglobin structure, this gives rise to abnormal hemoglobins.
The term 'thalassemia' describes a cluster of hemoglobin synthesis disorders, which are autosomal recessive conditions. The nomenclature is led by the location of the defect. α- and β-thalassemias have the greatest clinical significance. Homozygous forms of thalassemias are accompanied by serious, hypochromic hemolytic anemias and complex diseases. Heterozygous carriers usually show a mild, iron-refractory, microcytic and hypochromic anemia. α-thalassemias are caused by an α-chain synthesis defect. At the molecular level, they result from partial (α+) or total (α0) deletions, or more rarely mutations of one or more of the four α-globin genes (αα/αα). They occur mainly in Africa, Arab nations, and, more frequently,
South-East Asia [34]. Accordingly, β-thalassemia syndromes are the result of insufficient (β+) or absent (β0) production of β-chains caused by β-globin gene mutations. Most people with β-thalassemia syndromes come from Mediterranean countries, South-East Europe, Arab nations, and Asia [34]. With respect to HbA1c determination, two confounding effects of thalassemias should be considered: the alteration of the hemoglobin molecule as analytical target and the potential impact of anemia.
The other group of hemoglobinopathies, abnormal hemoglobin variants, is autosomal dominant inherited hemoglobin disorders characterized by structural defects, resulting from an altered amino acid sequence in the α- or β-chains. While some of these hemoglobin variants are clinically harmless, others can cause illness. These latter are divided into the following four well-defined groups [35]:
- Variants with a tendency to aggregate and with sickle cell formation (e.g., the sickle cell hemoglobin HbS [36];
- Variants with abnormal hemoglobin synthesis (e.g., HbE) [37];
- Variants with a tendency to precipitate and with hemolysis (e.g., Hb Köln) [38];
- Variants with abnormal oxygen transport and congenital polycythemia (e.g., Hb Johnstown) [39].
Variants of the third and fourth group cause serious illness when heterozygous, and can be fatal when in homozygous form.
Correct interpretation of HbA1c measurements depends on normal erythrocyte lifespan. In individuals with sickle cell, HbC, or HbD disease it is, therefore, recommended to use other tests than HbA1c for the determination of glycemic control, such as glycated serum albumin. As heterozygous carriers show normal erythrocyte survival, HbA1c can be used as long as the hemoglobin variant neither interferes with the assay method itself, nor with glucose binding to hemoglobin. In addition, the presence of some variants can modify the net charge of the hemoglobin and/or the recognition of the glycated N-terminus by antibodies, resulting in inaccurate HbA1c concentrations for some methods. Therefore, the effect of each variant must be specifically examined with each HbA1c method. On its website, the NGSP provides a comprehensive overview of interferences for most of the commonly used HbA1c methods for the most common hemoglobin variants and derivatives [40].
With respect to interference of the most common hemoglobin variants with HbA1c determination, four different methods should be considered:
1. HbA1c-specific immunoassays: Antibodies commonly recognize a structure of 4 to 10 amino acids at the N-terminus of the β-chain including the glycated N- terminal valine. Some, but not all, of these methods are affected by the presence of HbS and HbC variants, as the underlying mutations of the β-chain are close to the N- terminus [50]. In contrast, the presence of HbE or HbD with mutations much further away on the β-chain usually does not affect antibody-based methods [41].
2. Ion-exchange HPLC: Separation of hemoglobin molecules is based on charge differences between HbA1c and other hemoglobins. As the amino acid modifications in hemoglobin variants S, C, D, and E cause a change in the net charge of the hemoglobin molecule, these methods may cause interference [41,42]. In some cases the hemoglobin variant may coelute with HbA1c. Fortunately, interferences from hemoglobin variants using ion-exchange HPLC methods can often be detected in the chromatograms (Fig. 5).
HPLC chromatogram showing a HbE pattern hemoglobinopathy interfering with the analysis of HbA1c.
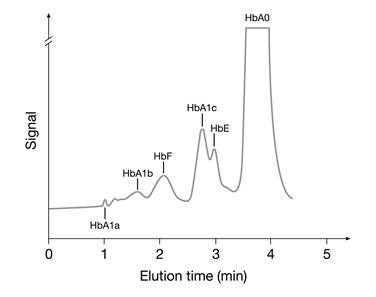
3. Boronate affinity methods: m-aminophenylboronic acid reacts specifically with the cis-diol groups of glucose bound to hemoglobin. Hence, boronate affinity measures the ratio of total glycated to non-glycated hemoglobin regardless of hemoglobin species. Therefore, this method tends to show the least interference from the presence of hemoglobin variants [41,42]. However, interference from elevated concentrations of HbF has been observed and is thought to be a consequence of a lower glycation rate for HbF compared with HbA. As a consequence, the presence of elevated HbF indicates a falsely too low HbA1c concentration [42].
4. Direct enzymatic method: This assay measures HbA1c by using an enzyme that specifically cleaves the N-terminal valine. Currently, there is only one assay available (Direct Enzymatic HbA1c Assay™, Diazyme Laboratories, Poway, CA, U.S.). No interferences due to the presence of HbS, C, D, E or elevated concentrations of HbF have been reported.
Regardless of the method used, any condition that shortens red blood cell survival or decreases mean erythrocyte age will falsely lower HbA1c. HbA1c results from people with diabetes with HbSS, HbCC, and HbSC must be interpreted with caution given the pathological processes, including anemia, increased red cell turnover, and transfusion requirements that adversely impact HbA1c as a marker of long-term glycemic control. For these people with diabetes, alternative forms of testing, such as glycated serum albumin, should be considered. Glycated serum albumin indicates average glucose levels over a much shorter period of time than the HbA1c test, usually about two or three weeks. However, the glycated serum albumin test is not standardized and the relationship of results of this test to glucose levels or risk for complications has not been established [43].
There are other factors in addition to the above-mentioned ones that have an influence on HbA1c. For reasons not yet understood, iron deficiency anemia can increase HbA1c concentrations by up to two percentage points; a process that is reversible when iron deficiency anemia is cured [44]. Conversely, pregnancy leads to a reduction in HbA1c concentrations. In an Italian multicenter study, the HbA1c reference intervals were 4.0%-5.5% (20-37 mmol/mol) for pregnant non-diabetic women and 4.8%-6.2% (29-44 mmol/mol) for non-pregnant controls [45].
As discussed earlier, HbA1c is an inappropriate marker for detecting rapid glucose changes and periods of acute hyper- or hypoglycemia. However, there is increasing evidence that especially these acute phases contribute to the common complications in diabetes.
What are the consequences of acute hyperglycemia in critically ill patients?
In patients experiencing an acute phase of critical illness such as severe brain injury [46], trauma [47,48], myocardial infarction [49] or stroke [50], hyperglycemia and insulin resistance are commonly observed, even if these patients have not previously been diagnosed with diabetes [51-53].
In numerous different clinical settings with critically ill patients [46-50], elevated glucose levels, even at modest degrees, have been identified as an independent risk factor for substantial increase in in-hospital morbidity [54-56] and mortality [54,55,57]. In 2001, data from the landmark clinical trial conducted by Van den Berghe et al. [54] showed that tight glycemic control with intensive insulin therapy, targeting achievement, and maintenance of normoglycemia (approximately 90-99 mg/dL [5.0-5.5 mmol/L] mean blood glucose levels) significantly lowered mortality and prevented several most feared complications such as bloodstream infections, acute renal failure, bacteremia, and polyneuropathy associated with critical illness.
In striking contrast to the findings by Van den Berghe et al. [54], investigators of the Normoglycaemia in Intensive Care Evaluation-Survival Using Glucose Algorithm Regulation (NICE-SUGAR) [58] trial concluded from their results that a blood glucose target of less than 180 mg/dL (10.0 mmol/L) resulted in lower mortality than a target of 81-108 mg/dL (4.5-6.0 mmol/L). Their findings suggested that a goal of normoglycemia for glucose control is not necessarily beneficial to critically ill patients and may be harmful, thus not recommending use of the lower target in critically ill adults. Conversely, several other studies [56,59-61] evaluating tight blood glucose control in populations on medical and surgical intensive care units (ICUs) revealed comparable beneficial findings regarding both mortality and morbidity comparable to the Van den Berghe et al. [54] findings, even though the mechanisms by which the intensive insulin therapy improves outcomes are not completely elucidated.
During the acute phase of physical and mental stress in critically ill patients, the endocrine stress response including elevated levels of cytokines, growth hormone, glucagon, and cortisol opposes the normal action of insulin and increases lipolysis and proteolysis, providing substrates for the hepatic gluconeogenesis. Supportingly, released catecholamines enhance hepatic glycogenolysis and inhibit glycogenesis, resulting in elevated blood glucose levels despite increased levels of released insulin [62]. The increased levels of released insulin, impaired peripheral glucose uptake, and elevated hepatic glucose production reflect the development of insulin resistance [63], constituting the vicious circle of hyperglycemia that must be addressed.
When treatment with insulin is initiated, it can be assumed that the insulin therapy lowers glucose levels mainly via increase of insulin-stimulated glucose uptake and intracellular glucose metabolism in skeletal muscle, heart, and adipose tissue by means of up-regulation of glucose carrier and transporter density as well as intracellular enzyme activity [64]. Furthermore, intensive insulin therapy prevents excessive inflammation [65,66], restores leukocyte function [67], and stimulates the anti-inflammatory cascade [68], compensating for increased risk of infection caused by hyperglycemia.
Does chronic hyperglycemia in people with diabetes have the same consequences as in critically ill patients?
The exposure to elevated blood glucose levels in people with diabetes involves two components: the duration and magnitude of chronically sustained hyperglycemia, reflected in HbA1c, and the acute fluctuations of glucose around a mean value from peaks to nadirs over a daily period [69-73], reflecting intermittent acute glucose toxicity, obviously illustrated by glycemic variability. As a parameter for the overall glycemic control, HbA1c thus reveals little about individual daily glucose fluctuations [74].
In vitro and in vivo studies have shown a significantly more deleterious effect of oscillating glucose levels over a 24-h period to endothelial function than stable constant high glucose levels by activating oxidative stress pathways [75,76], significantly contributing to promoting lipid peroxidation and decreasing antioxidation capacity [77,78]. Dysfunction of the vascular endothelium may be one of the most critical inducers of micro- and macrovascular damages in diabetes [79-81]. A growing body of evidence indicates that recurrent and/or periodic blood glucose fluctuations with large amplitude levels beyond near-normoglycemic limits play a much more serious role in diabetic vascular damage than chronically sustained hyperglycemia.
Is lowering of HbA1c the key to reduce cardiovascular risk?
Although microvascular complications including nephropathy, retinopathy, and neuropathy can lead to significant morbidity and premature mortality, the greatest cause of death in people with diabetes is cardiovascular disease (CVD); the risk for CVD in people with diabetes is increased two- to fourfold compared to people without diabetes [82,83-85]. Until recently, there has been little evidence that specifically targeting glycemic control can reduce the frequency of cardiovascular end-points [86,87].
Several large epidemiological prospective trials, partly with long-term results, have been conducted to determine whether intensive glycemic control prevents microvascular complications, cardiovascular events, and mortality in people with diabetes [88]. Table 1 summarizes their findings about risk reduction of CVD owing to tight glycemic control.
Clinical trials investigating the reduction of cardiovascular risk under tight glycemic control.
| Trials/ Diabetes mellitus type | Mean age (years) | Mean diabetes duration at baseline (years) | Mean treat- ment duration (years) | Treatment | Target FBG/ Target pre- prandial BG Target PPBG | Target HbA1c [% (mmol/mol)] | Mean HbA1c achieved [% (mmol/mol)] | Risk reduction of macrovascular events/ disease |
|---|---|---|---|---|---|---|---|---|
| DCCT [2] / T1DM | ST group: 27 ± 7; IT group: 27 ± 7. | ST group: 5 ± 4; IT group: 6 ± 4. | 6.5 | ST group: CT; IT group: MDI or IPT. | ST group: no targets defined; IT group: pre- prandial BG: 70 < BG <120 mg/dL (3.9 < BG < 6.7 mmol/L); PPBG: < 180 mg/dL (< 10.0 mmol/L). | ST group: no target defined; IT group: < 6.05 (43). | ST group: 9.1 (76); IT group: 7.4 (57). | ST group vs. IT group: major cardiovascular and peripheral vascular events by 41% in IT group; no statistical significance between ST group and IT group. |
| EDIC [87,88] (observatio nal long- term follow- up study of DCCT) / T1DM | ST group: 33 ± 7; IT group: 34 ± 7. | ST group: 12 ± 5; IT group: 12 ± 5. | 11 | ST group: CT; IT group: MDI or IPT. | ST group: no targets defined; IT group: no targets defined; | ST group: no target defined; IT group: no target defined. | ST group: 8.2 (66); IT group: 8.0 (64). | ST group vs. IT group: IT group: by 42% for any cardiovascular event, by 57% for non-fatal myocardial infarction, stroke, or death from cardiovascular disease in the IT group. |
| UKPDS [89] / T2DM | ST group: 53 ± 9; IT group: 53 ± 9. | ST group: newly diagnosed; IT group: newly diagnosed. | 11.1 | ST group: diet alone, by need combined with insulin, oral antidiabetics; IT group: diet + oral antidiabetics, by need insulin (dose adjust. with SMBG). | ST group: FBG < 270 mg/dL (< 15.0 mmol/L); IT group: FBG < 108 mg/dL (< 6.0 mmol/L), in insulin- treated pat.: pre- prandial BG: 72- 126 mg/dL (4.0-7.0 mmol/L). | ST group: no target defined; IT group: no target defined. | ST group: 7.9 (63); IT group: 7.0 (53). | ST group vs. IT group: by 12% for microvascular and macrovascular events in the IT group; ST group vs. IT group: no statistically significant reduction for diabetes-related mortality, all- cause mortality, non-fatal myocardial infarction, stroke, cardiomegaly, and peripheral vascular disease. |
| UKPDS Post-Trial Monitoring [88,90] (10-year, post- intervention al follow-up of the UKPDS survivor cohort) / T2DM | ST group: 63 ± 9; IT group: 63 ± 9. | ST group: 11.1; IT group: 11.1. | 16.8 - 17.7 | ST group: diet alone, by need combined with insulin, oral antidiabetics; IT group: diet +oral antidiabetics, by need insulin (dose adjust. with SMBG). | ST group: no targets defined; IT group: no targets defined. | ST group: no target defined; IT group: no target defined. | ST group: 7.8 (62); IT group: 7.8 (62). | ST group vs. IT group: by 15% for myocardial infarction, by 13% for death from any cause in the IT group. |
| ACCORD [88,91] / T2DM | ST group: 62 ± 7; IT group: 62 ± 7. | ST group: 10; IT group: 10. | 3.5 | Any therapeutic regimens with any marketed antihyperglycemics. ST group: glycemic- management visits every 4 mos. IT group: monthly visits in first 4 mos., then every 2 mos. | ST group: no targets defined; IT group: no targets defined. | ST group: 7.0 - 7.9 (53 - 63); IT group: < 6.0 (42). | ST group: 7.5 (59); IT group: 6.4 (47). | ST group vs. IT group: increased by 2.6% in the ST group, increased by 1.8% for death from cardiovascular causes. |
| ADVANCE [88,92,93] / T2DM | ST group: 66 ± 6; IT group: 66 ± 6. | ST group: 8.0 ± 6.4; IT group: 7.9 ± 6.3. | 5 | ST group: any medication but gliclizide. IT group: sulfonylurea, gliclizide and additional medications as needed. | ST group: no targets defined; IT group: no targets defined. | ST group: set accord. to “local guidelines; IT group: ≤ 6.5 (48). | ST group: 7.3 (56); IT group: 6.5 (48). | ST group vs. IT group: no statistically significant reduction for macrovascular events in the IT group. |
| VADT [86, 88, 94] / T2DM | ST group: 60 ± 9; IT group: 61 ± 9. | ST group: 11.5 ± 7.0; IT group: 11.5 ± 8.0. | 5.6 | ST group: 50% of max. doses of rosiglitazone plus glimepiride or metformin; IT group: max. doses of rosiglitazone plus glimepiride or metformin. | ST group: no targets defined; IT group: no targets defined. | ST group: no target defined; IT group: a 1.5% reduction beyond that achieved by ST group. | ST group: 8.4 (68); IT group: 6.9 (52). | ST group vs. IT group: no statistically significant reduction for macrovascular events in the IT group. |
Abbreviations in this table:
DCCT = Diabetes Control and Complications Trial
EDIC = Epidemiology of Diabetes Interventions and Complications, long-term follow-up study of DCCT UKPDS = United Kingdom Prospective Diabetes Study
ACCORD = Action to Control Cardiovascular Risk in Diabetes
ADVANCE = Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation
VADT = Veterans Affairs Diabetes Trial
ST= standard therapy
IT = intensive therapy
FBG = fasting blood glucose
preprandial BG= preprandial blood glucose
PPBG = postprandial blood glucose T1DM = Type 1 diabetes mellitus T2DM = Type 2 diabetes mellitus MDI = multiple daily insulin injection mos = months
IPT = insulin pump therapy
CT = conventional insulin therapy
Interestingly, a post hoc subgroup analysis of the Veterans Affairs Diabetes Trial (VADT) [95] suggested that people with a diabetes duration of less than 12 years appear to derive a cardiovascular benefit from intensive glycemic control. If diabetes had existed for more than 12 years at the time of study entry, however, cardiovascular event rates were either unchanged or even increased in people with diabetes treated with intensive glycemic control [95].
For the prevention of microvascular and macrovascular disease in people with diabetes, the currently recommended target of HbA1c concentrations should remain less than 7.0% (53 mmol/mol) [96], particularly in young people with T1DM and in individuals with newly diagnosed type 2 diabetes (T2DM) [93]. Less stringent HbA1c goals of greater than 7.0% (53 mmol/mol) might be indicated for people with type 2 diabetes who have extensive comorbid conditions, limited life expectancy, or an increased risk of severe hypoglycemia. Considering that hypoglycemia was associated with an increased risk of cardiovascular events in both the intensive therapy and standard treatment, substantial hypoglycemia should be avoided [82].
As demonstrated in the trials Action to Control Cardiovascular Risk in Diabetes (ACCORD), Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE), and VADT, a target HbA1c concentration of ≤ 6.5% (48 mmol/mol) (near-normal range) was unable to show reduction of macrovascular complications in people with type 2 diabetes.
In people with T1DM, a period of intensified glycemic control results in a subsequent risk reduction of any cardiovascular event, non-fatal myocardial infarction, stroke, or death from CVD many years after the initial intervention [86,87,93]. This late cardiovascular benefit has been described as the legacy effect and is believed to confer a so-called metabolic memory or long-term cardiovascular protection [87,88,97]. However, the pathophysiological mechanisms responsible for it remain unclarified.
It has been postulated that people with T2DM would also realize long-term cardiovascular protection through early and intensive control of glycemia, resulting in a significant reduction in myocardial infarctions and total mortality [86]. However, in people with a long history of T2DM and high cardiovascular risk, intensive glucose control reduces the rate of events in coronary heart disease and CVD but not cardiovascular or total mortality [86,90,93,97,98].
As reported in the DCCT´s long-term follow-up study Epidemiology of Diabetes Interventions and Complications (EDIC) [87] and, particularly, in the UKPDS Post-Trial Monitoring [90] data analysis, the difference in HbA1c concentrations between the intensive and conventional therapy groups was lost over time. This phenomenon was observed in the latter only one year after the UKPDS closed. As both the intensive and conventional therapy resulted in comparable HbA1c concentrations persisting over time, the observed legacy effect under intensive treatment is, therefore, highly unlikely derived only from lowered HbA1c concentrations but assumingly also from additional factors.
Reduction of glycemic variability by means of SMBG - does it make the difference?
In the ACCORD study, people with diabetes in the intensive-therapy group attended monthly visits for the first four months and then every two months thereafter with the aim of rapidly and safely reducing HbA1c concentrations to below 6.0% (42 mmol/mol) [99,100]. The study protocol mentioned that people with diabetes not willing to do frequent capillary blood glucose self-monitoring were excluded.
On the basis of the glycated hemoglobin concentration at each visit, the ADVANCE trial protocol initially advised increasing the dose with the sequential addition of oral antidiabetics or insulin with the initial use of basal insulin and addition of short-acting insulin at meals for people with diabetes in whom the target HbA1c concentration was not achieved [91].
In the VADT study, insulin was added for people with diabetes in the intensive-therapy group who did not achieve an HbA1c concentration of less than
6.0% (42 mmol/mol) and for those in the standard-therapy group with a concentration of less than 9.0% (75 mmol/mol) before any change in oral medications. Subsequent changes in medication were determined according to protocol guidelines and local assessment. The goal for the HbA1c concentrations was an absolute reduction of 1.5 percentage points in the intensive-therapy group as compared with the standard-therapy group [92].
None of these epidemiological trials, ACCORD, ADVANCE or VADT, integrated self-monitoring of blood glucose (SMBG) with documented glucose levels obtained at prescribed preprandial and postprandial time points over a daily period to determine dosage adjustments of antidiabetic agents in their study procedures. Use of this type of “structured” SMBG regimen would have shown blood glucose profiles over a daily period, providing fundamental information about the potential need for differentiated dosage adjustments of preprandially and/or postprandially effective hypoglycemic agents. Instead, the dosage adjustment of antidiabetic oral agents or insulins was determined by the set target HbA1c concentrations, not by the preprandial and/or postprandial blood glucose levels.
In contrast to the ACCORD, ADVANCE and VADT trials, the treatment adjustment practice integrated in the DCCT, UKPDS and the most recent STeP (Structured Testing Program) study [101,102] was based on the measured glucose levels at several points of time predefined during the day.
The aim of intensive treatment in the UKPDS was FPG less than 108 mg/dL (6.0 mmol/L) and, in insulin-treated people with diabetes, pre-meal glucose concentrations of 72-126 mg/dL (4.0-7.0 mmol/L). Whenever glucose concentrations were above target concentrations, a letter was sent from the coordinating center with advice on necessary changes in therapy. Insulin-treated subjects started on once daily ultralente insulin or isophane insulin. If the daily dose was >14 units (U) or pre-meal or bed-time SMBG measurements were >126 mg/dL (7.0 mmol/L), a short-acting insulin, usually soluble regular insulin, was added, i.e., basal/bolus regimen. Subjects on more than 14 U insulin per day or on short-acting insulins were particularly encouraged to do regular SMBG [89].
In the DCCT [2] with people with T1DM, intensive therapy included the administration of insulin three or more times daily by injection or an external pump. The dosage was adjusted according to the results of SMBG performed at least four times per day, dietary intake, and anticipated exercise. Blood glucose concentrations achieved with each treatment arm (intensive therapy and conventional therapy) were measured with quarterly seven-point capillary-blood glucose profiles. The mean value for all glucose profiles in the intensive therapy group was 155 mg/dL (8.6 mmol/L), as compared with 231 mg/dL (12.8 mmol/L) in the conventional therapy group, i.e., in the intensive therapy group, seven-point capillary-blood glucose values including preprandial and postprandial glucose excursions were shifted down towards the normoglycemic range, reducing excursion amplitudes lying far beyond near-normal range. The differences between treatments were statistically significant at each of the seven testing times [2].
Most recently, Polonsky et al. [101,102] published the STeP study results, assessing the effectiveness of structured blood glucose testing in poorly controlled, non-insulin-treated T2DM. People with diabetes with a duration of T2DM for more than 1 year, aged ≥ 25 years and HbA1c concentrations ≥ 7.5% (59 mmol/mol) to < 12.0% (108 mmol/mol) were included. The primary end point was a change in HbA1c from screening to 12 months in subjects using structured SMBG in conjunction with enhanced usual care (structured testing group [STG]) compared to an active control group (ACG) that received enhanced usual care only. Enhanced usual care included quarterly clinic visits that focused specifically on diabetes management, free blood glucose meters and strips, and office point-of-care HbA1c capability.
STG participants used the validated Accu-Chek® 360° View 3-day profile tool [103], a validated paper tool, that enabled people with diabetes to record and plot a seven-point SMBG profile (fasting, preprandial/2-h postprandial at each meal, bedtime) on three consecutive days prior to each scheduled study visit.
STG physicians/staff received training on interpreting the structured data and were provided with an algorithm that described various pharmacologic (initiation of a new medication, increase/decrease in the dose of an existing medication, and/ or termination of an existing medication) and lifestyle (defined as any change in diet, exercise, or other self-care behavior) treatment strategies that could be used in response to the specific SMBG patterns identified.
Both the intent-to-treat (ITT) and per protocol (PP) analyses revealed that both groups showed significant reductions in HbA1c concentrations. However, STG subjects in the intent-to-treat group evidenced significantly greater mean reductions in HbA1c than ACG subjects over the 12 months (-1.2% vs. -0.9%). Per protocol analysis revealed an even greater mean HbA1c reduction among those STG subjects who adhered to the intervention compared with ACG subjects (-1.3% vs. -0.8%) [102]. Early treatment modification recommendations (TMRs) were associated with significantly greater glycemic improvement than later treatment modification recommendations, especially in people with diabetes with the poorest glycemic control at baseline (HbA1c ≥ 8.5% [70 mmol/mol]) [104].
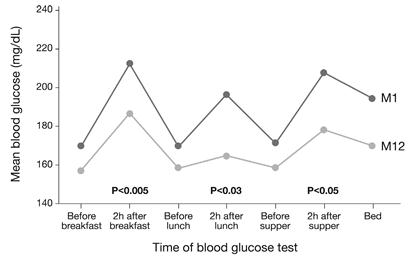
Furthermore, STG subjects showed significantly lower average preprandial and postprandial glucose levels at all meals and at bedtime from month 1 to month 12 in all cases. More importantly, there was a significant drop from month 1 to month 12 in preprandial to postprandial glucose excursions at all meals (Fig. 6) [105]. Measurements of mean amplitude of glucose excursions indicated significant mean reductions in glycemic variability among STG subjects from 38.5 mg/dL at month 1 to 34.3 mg/dL at month 12 [102], thus shifted down towards near-normoglycemic range.
In the Structured Testing Program (STeP) study [108], people with diabetes in the structured testing group showed significant reductions in average, preprandial, postprandial and bedtime blood glucose levels at Month 12 (p<0.01). The degree of glycemic excursions at breakfast, lunch and supper was also significantly improved (intent-to-treat analysis). Abbr.: M1 (Month 1), M12 (Month 12).
In contrast to conflicting data about effectiveness of SMBG from other trials [106-114], the STeP study results were able to demonstrate that using pattern recognition of blood glucose levels by means of SMBG profiles and subsequent early treatment modifications by involving physicians or other healthcare professionals lead to significant reduction of HbA1c, and simultaneously, of glycemic variability in the study participants.
Although long-term outcome data are unavailable in the STeP study due to the lack of time potentially showing the legacy effect in the study subjects, the results in the DCCT´s long-term follow-up study EDIC [87] and particularly UKPDS Post-Trial Monitoring [90] about cardiovascular risk reduction confirm the existence of this late beneficial cardiovascular protection effect. At the cellular and molecular level, it is plausible that reducing glucose oscillations back into near-normoglycemic limits might arrest or at least decelerate the progression of diabetic vascular damage by slowing the gradual accumulation of advanced glycation end products that are subsequently degraded with intensive glycemic control leading to the legacy effect. The results recently published in a systematic review [115] on a total of 18 studies (8 on T1DM and 10 on T2DM patients) showed that there is increasing evidence indicating a possible link between glycemic variability and the risk of developing both micro- and macrovascular complications as well as mortality among patients with T2DM. In contrast, such a relationship, however, has not been observed in studies evaluating patients with T1DM. In diabetic patients with T2DM, both pre- and postprandial glucose (PPG) peaks seem to promote the development of long-term cardiovascular complications, independently of fasting plasma glucose (FPG) and HbA1c values. The authors of this systematic review also concluded that glycemic variability - in addition to HbA1c levels - should be considered as a target of diabetic therapy.
In favor of reducing the progression of CVD risk, the best possible benefit from lowering HbA1c levels to the established HbA1c targets [96,105] in people with T1DM and T2DM derives from the well-known legacy effect that might potentially be strengthened over time by reducing glycemic variability with conduction of SMBG.
Conclusion
Since its adoption in 1993 with the DCCT, the measurement of HbA1c as a marker for monitoring the glycemic status of people with diabetes has reached such a high level of analytical quality that it is currently suggested also to be used for the diagnosis of diabetes. Nevertheless, it is crucial to be aware of possible interferences during its measurement, depending on many factors including ethnic differences among people with diabetes, pathological conditions such as hemoglobinopathies, and the methods used to determine HbA1c concentrations. However, discussions about the clinical relevance of HbA1c regarding risk reduction of cardiovascular morbidity and mortality are ongoing. Beside well-established risk factors for CVD, such as hypertension or hyperlipidemia, glycemic variability as a further risk factor should deserve more attention. As shown by available clinical and non-clinical data, lowering HbA1c concentrations combined with reducing glycemic variability by means of SMBG profiles very soon after the diagnosis of diabetes will likely result in the greater benefit with regard to CVD risk reduction. Future prospective trials assessing the effect of the reduction of glycemic variability on the development of long-term diabetic micro- and macrovascular complications are needed to further strengthen currently available results.
Abbreviations
A1c: Hemoglobin A1c; ACCORD: Action to Control Cardiovascular Risk in Diabetes; ACG: active control group; ADA: American Diabetes Association; ADAG: A1c- Derived Average Glucose; ADVANCE: Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation; AG: average glucose; CAP: College of American Pathologists; CVD: cardiovascular disease; CT: conventional insulin therapy; DCCT: Diabetes Control and Complications Trial; EASD: European Association for the Study of Diabetes; EDIC: Epidemiology of Diabetes Interventions and Complications, long-term follow-up study of DCCT; FPG: fasting plasma glucose; ICU: intensive care unit; IDF: International Diabetes Federation; IFCC: International Federation for Clinical Chemistry and Laboratory Medicine; IGT: impaired glucose tolerance; IPT: insulin pump therapy; ISPAD: International Society for Pediatric and Adolescent Diabetes; ITT: intent-to-treat; IT: intensive therapy; GLUT: glucose transporter; Hb: Hemoglobin; HbA1c: Hemoglobin A1c; ; HbAC: HbC trait; HbAD: HbD trait; HbAE: HbE trait; HbAS: sickle cell trait; HPLC: high performance liquid chromatography; MDI: multiple daily insulin injection; NDDG: National Diabetes Data Group; NGSP: National Glycohemoglobin Standardization Program; NHANES: National Health and Nutrition Examination Survey; NICE-SUGAR: Normoglycaemia in Intensive Care Evaluation-Survival Using Glucose Algorithm Regulation; NPDR: nonproliferative diabetic retinopathy; OGTT: oral glucose tolerance test; PG: plasma glucose; PP: per protocol; PPBG: postprandial blood glucose; preprandial BG: preprandial blood glucose; SMBG: self- monitoring of blood glucose; STeP: Structured testing Program; STG: structured testing group; ST: standard therapy; T1DM: Type 1 diabetes mellitus; T2DM: Type 2 diabetes mellitus; TMR: treatment modification recommendation; U: unit; UKPDS: United Kingdom Prospective Diabetes Study; VADT: Veterans Affairs Diabetes Trial; WHO: World Health Organization.
References
1. Rabhar S, Blumenfeld O, Ranney H. Studies of an unusual hemoglobin in patients with diabetes mellitus. Biochem Biophys Res Commun. 1969;36(5):838-43
2. Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long- term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329(14):977-86
3. Goodall I. HbA1c standardisation. Destination - global IFCC standardisation. How, why, where and when. Clin Biochem Rev. 2005;26(1):5-20
4. John WG, Mosca A, Weykamp C, Goodall I. HbA1c standardisation: history, science and politics. Clin Biochem Rev. 2007;28(4):163-8
5. Little R. Glycated hemoglobin standardization - National Glycohemoglobin Standardization Program (NGSP) perspective. Clin Chem Lab Med. 2003;41(9):1191-8
6. National Glycohemoglobin Standardization Program. http://www.ngsp.org
7. Hoelzel W, Miedema K. Development of a reference system for the international standardization of HbA1c/glycohemoglobin determinations. J Int Fed Clin Chem. 1996;9(2):66-7
8. Finke A, Kobold U, Hoelzel W, Weykamp C, Miedema K, Jeppsson J. Preparation of a candidate primary reference material for the international standardisation of HbA1c determinations. Clin Chem Lab Med. 1998;36(5):299-308
9. Jeppsson JO, Kobold U, Barr J, Finke A, Hoelzel W, Hoshino T, Miedema K, Mosca A, Mauri P, Paroni R, Thienpont L, Umemoto M, Weykamp C; International Federation of Clinical Chemistry, Laboratory medicine (IFCC). Approved IFCC reference method for the measurement of HbA1c in human blood. Clin Chem Lab Med. 2002;40(1):78-89
10. International Federation of Clinical Chemistry and Laboratory Medicine. http://www.ifcchba1c.net
11. Hanas R, John G; International HbA1c Consensus Committee. 2010 consensus statement on the worldwide standardization of the hemoglobin A1c measurement. Diabetes Care. 2010;33(8):1903-4
12. Little R, Rohlfing C. HbA1c. An overview of current analytical testing issues. Clin Lab News. 2011;37(2):8-10
13. Al-Ansary L, Farmer A, Hirst J, Roberts N, Glasziou P, Perera R, Price C. Point-of-care testing for Hb A1c in the management of diabetes: a systematic review and metaanalysis. Clin Chem. 2011;57(4):568-76
14. National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes. 1979;28(12):1039-57
15. Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20(7):1183-97
16. Colagiuri S, Borch-Johnsen K. Detect-2: early detection of type 2 diabetes and IGT. Diabetes Voice. 2003;48(4):11-3
17. International Expert Committee. International Expert Committee Report on the Role of the A1C Assay in the Diagnosis of Diabetes. Diabetes Care. 2009;32(7):1327-34
18. Selvin E, Crainiceanu C, Brancati F, Coresh J. Short-term variability in measures of glycemia and implications for the classification of diabetes. Arch Intern Med. 2007;167(14):1545-51
19. Bao Y, Ma X, Li H, Zhou M, Hu C, Wu H, Tang J, Hou X, Xiang K, Jia W. Glycated haemoglobin A1c for diagnosing diabetes in Chinese population: cross sectional epidemiological survey. Diabetes Care. 2010;33(9):2184-9
20. Pinelli N, Jantz A, Martin E, Jaber L. Sensitivity and specificity of glycated hemoglobin as a diagnostic test for diabetes and prediabetes in arabs. J Clin Endocrinol Metab. 2011;96(10):E1680-3
21. Olson D, Rhee M, Herrick K, Ziemer D, Twombly J, Phillips L. Screening for diabetes and pre-diabetes with proposed A1C-based diagnostic criteria. Diabetes Care. 2010;33(10):2184-9
22. Day A. HbA1c and diagnosis of diabetes. The test has finally come of age. Ann Clin Biochem. 2012;49(Pt 1):7-8
23. Nathan D, Kuenen J, Borg R, Zheng H, Schoenfeld D, Heine R. Translating the A1c assay into estimated average glucose values. Diabetes Care. 2008;31(8):1473-8
24. Herman W, Cohen R. Racial and ethnic differences in the relationship between HbA1c and blood glucose: Implications for the diagnosis of diabetes. J Clin Endocrinol Metab. 2012;97(4):1067-72
25. Riddle M, Umpierrez G, Digenio A, Zhou R, Rosenstock J. Contributions of basal and postprandial hyperglycemia over a wide range of A1C levels before and after treatment intensification in type 2 diabetes. Diabetes Care. 2011;34(12):2508-14
26. Carruthers A. Facilitated diffusion of glucose. Physiol Rev. 1990;70(4):1135-1176
27. Khera P, Joiner C, Carruthers A, Lindsell C, Smith E, Franco R, Holmes Y, Cohen R. Evidence for interindividual heterogeneity in the glucose gradient across the human red blood cell membrane and its relationship to hemoglobin glycation. Diabetes. 2008;57(9):2445-52
28. Cohen R, Holmes Y, Chenier T, Joiner C. Discordance between A1C and fructosamine: evidence for a glycosylation gap and its relation to diabetic nephropathy. Diabetes Care. 2003;26(1):163-7
29. Saaddine J, Fagot-Campagna A, Rolka D, Narayan K, Geiss L, Eberhardt M, Flegal K. Distribution of HbA(1c) levels for children and young adults in the U.S.: Third National Health and Nutrition Examination Study. Diabetes Care. 2002;25(8):1326-30
30. Herman W. Do race and ethnicity impact hemoglobin A1c independent of glycemia? J Diabetes Sci Technol. 2009;3(4):656-60
31. Cohen R, Snieder H, Lindsell C, Beyan H, Hawa M, Blinko S, Edwards R, Spector T, Leslie R. Evidence for independent heritability of the glycation gap (glycosylation gap) fraction of HbA1c in nondiabetic twins. Diabetes Care. 2006;29(8):1739-43
32. Virtue M, Furne J, Nuttall F, Levitt M. Relationship between GHb concentration and erythrocyte survival determined from breath carbon monoxide concentration. Diabetes Care. 2004;27(4):931-5
33. Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization. http://www.who.int/bulletin/volumes/86/6/06-036673/en/
34. Weatherall D, Clegg J. The thalassaemia syndromes, 4th ed. Oxford, UK: Blackwell Science Ltd. 2001
35. Kohne E. Hemoglobinopathies: Clinical Manifestations, Diagnosis, and Treatment. Dtsch Arztebl Int. 2011;108(31-32):532-40
36. Steinberg M. Sickle cell anemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. ScientificWorldJournal. 2008;8:1295-324
37. Weatherall D, Clegg J, Higgs D, Wood W. The hemoglobinopathies. In: (ed.) Scriver C, Beauder A, Sly W, Valle D. The metabolic basis of inheited disease, 8th ed. New York: McGraw-Hill. 2000:4571-636
38. Ohba Y. Unstable hemoglobins. Hemoglobin. 1990;14(4):353-88
39. Wajcman H, Galactéros F. Hemoglobins with high oxygen affinity leading to erythrocytosis, new variants and new concepts. Hemoglobin. 2005;29(2):91-106
40. Factors that interfere with HbA1c test results. National Glycohemoglobin Standardization Program 2010. http://www.ngsp.org/factors.asp
41. Little R, Rohlfing C, Hanson S, Connolly S, Higgins T, Weykamp C, D'Costa M, Luzzi V, Owen W, Roberts W. Effects of hemoglobin (Hb) E and HbD traits on measurements of glycated Hb (HbA1c) by 23 methods. Clin Chem. 2008;54(8):1277-82
42. Little R, Roberts W. A review of variant hemoglobins interfering with hemoglobin A1c measurement. J Diabetes Sci Technol. 2009;3(3):446-51
43. Sickle cell trait and other hemoglobinopathies and diabetes: important information for physicians. National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health (NIH). http://diabetes.niddk.nih.gov/dm/pubs/hemovari-A1C/index.aspx#A1CTest
44. Coban E, Ozdogan M, Timuragaoglu A. Effect of iron deficiency anemia on the levels of hemoglobin A1c in nondiabetic patients. Acta Haematol. 2004;112(3):126-8
45. Mosca A, Paleari R, Dalfrà M, Di Cianni G, Cuccuru I, Pellegrini G, Malloggi L, Bonomo M, Granata S, Ceriotti F, Castiglioni M, Songini M, Tocco G, Masin M, Plebani M, Lapolla A. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem. 2006;52(6):1138 -
46. Rovlias A, Kotsou S. The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery. 2000;46(2):335-42
47. Yendamuri S, Fulda G, Tinkoff G. Admission hyperglycemia as a prognostic indicator in trauma. J Trauma. 2003;55(1):33-8
48. Bochicchio G, Sung J, Joshi M, Bochicchio K, Johnson S, Meyer W, Scalea T. Persistent hyperglycemia is predictive of outcome in critically ill trauma patients. J Trauma. 2005;58(5):921-4
49. Capes S, Hunt D, Malmberg K, Gerstein H. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355(9206):773-8
50. Capes S, Hunt D, Malmberg K, Pathak P, Gerstein H. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: a systematic overview. Stroke. 2001;32(10):2426-32
51. Wolfe R, Allsop J, Burke J. Glucose metabolism in man: responses to intravenous glucose infusion. Metabolism. 1979;28(3):210-20
52. Wolfe R, Herndon D, Jahoor F, Miyoshi H, Wolfe M. Effect of severe burn injury on substrate cycling by glucose and fatty acids. N Engl J Med. 1987;317(7):403-8
53. Shangraw R, Jahoor F, Miyoshi H, Neff W, Stuart C, Herndon D, Wolfe R. Differentiation between septic and postburn insulin resistance. Metabolism. 1989;38(10):983-9
54. Van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345(19):1359-67
55. Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters P, Milants I, Van Wijngaerden E, Bobbaers H, Bouillon R. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354(5):449-61
56. Grey N, Perdrizet G. Reduction of nosocomial infections in the surgical intensive-care unit by strict glycemic control. Endocr Pract. 2004;10(Suppl 2):46-52
57. Krinsley J. Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients. Mayo Clin Proc. 2003;78(12):1471-8
58. Finfer S, Chittock D, Su S, Blair D, Foster D, Dhingra V, Bellomo R, Cook D, Dodek P, Henderson W, Hébert P, Heritier S, Heyland D, McArthur C, McDonald E, Mitchell I, Myburgh J, Norton R, Potter J, Robinson B, Ronco J. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360(13):1283-97
59. Van den Berghe G, Schoonheydt K, Becx P, Bruyninckx F, Wouters P. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology. 2005;64(8):1348-53
60. Ingels C, Debaveye Y, Milants I, Buelens E, Peeraer A, Devriendt Y, Vanhoutte T, Van Damme A, Schetz M, Wouters P, Van den Berghe G. Strict blood glucose control with insulin during intensive care after cardiac surgery: impact on 4-years survival, dependency on medical care, and quality-of-life. Eur Heart J. 2006;27(22):2716-24
61. Krinsley J. Effect of an intensive glucose management protocol on the mortality of critically ill adult patients. Mayo Clin Proc. 2004;79(8):992-1000
62. Watt M, Howlett K, Febbraio M, Spriet L, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol. 2001;534(Pt 1):269-78
63. Langouche L, Vanhorebeek I, Van den Berghe G. Therapy insight: the effect of tight glycemic control in acute illness. Nat Clin Pract Endocrinol Metab. 2007;3(3):270-8
64. Mesotten D, Swinnen J, Vanderhoydonc F, Wouters P, Van den Berghe G. Contribution of circulating lipids to the improved outcome of critical illness by glycemic control with intensive insulin therapy. J Clin Endocrinol Metab. 2004;89(1):219-26
65. Hansen T, Thiel S, Wouters P, Christiansen J, Van den Berghe G. Intensive insulin therapy exerts antiinflammatory effects in critically ill patients and counteracts the adverse effect of low mannose-binding lectin levels. J Clin Endocrinol Metab. 2003;88(3):1082-8
66. Weekers F, Giulietti A, Michalaki M, Coopmans W, Van Herck E, Mathieu C, Van den Berghe G. Metabolic, endocrine, and immune effects of stress hyperglycemia in a rabbit model of prolonged critical illness. Endocrinology. 2003;144(12):5329-38
67. Ellger B, Debaveye Y, Vanhorebeek I, Langouche L, Giulietti A, Van Etten E, Herijgers P, Mathieu C, Van den Berghe G. Survival benefits of intensive insulin therapy in critical illness: impact of maintaining normoglycemia versus glycemia-independent actions of insulin. Diabetes. 2006;55(4):1096-105
68. Jeschke MG, Klein D, Herndon D. Insulin treatment improves the systemic inflammatory reaction to severe trauma. Ann Surg. 2004;239(4):553-60
69. Stratton I, Adler A, Neil H, Matthews D, Manley S, Cull C, Hadden D, Turner R, Holman R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321(7258):405-12
70. Diabetes Control and Complications Trial Research Group. The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes. 1995;44(8):968-83
71. Klein R. Hyperglycemia and microvascular and macrovascular disease in diabetes. Diabetes Care. 1995;18(2):258-68
72. Bonora E, Muggeo M. Postprandial blood glucose as a risk factor for cardiovascular disease in Type II diabetes: the epidemiological evidence. Diabetologia. 2001;44(12):2107-14
73. Ceriello A, Hanefeld M, Leiter L, Monnier L, Moses A, Owens D, Tajima N, Tuomilehto J. Postprandial glucose regulation and diabetic complications. Arch Intern Med. 2004;164(19):2090-5
74. Bode B. Defining the importance of daily glycemic control and implications for type 2 diabetes management. Postgrad Med. 2009;121(5):82-93
75. Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol J, Colette C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006;295(14):1681-7
76. Ceriello A, Esposito K, Piconi L, Ihnat M, Thorpe J, Testa R, Boemi M, Giugliano D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes. 2008;57(5):1349-54
77. Wang Z, Li L, Zheng F, Jia C, Ruan Y, Li H. Correlation between the amplitude of glucose excursion and the oxidative/antioxidative system in subjects with different types of glucose regulation. Biomed Environ Sci. 2011;24(1):68-73
78. Zheng F, Wang Z, Li H, Jia CF, Zhang N, Yuan H. Correlation between the amplitude of glucose excursion and plasma 8-iso prostaglandin F2alpha level in subjects with different types of glucose regulation. Zhonghua Yi Xue Za Zhi. 2009;89(10):651-4
79. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813-20
80. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615-25
81. Hammes H. Pathophysiological mechanisms of diabetic angiopathy. J Diabetes Complications. 2003;17(Suppl 2):16-9
82. Moghissi E, Korytkowski M, DiNardo M, Einhorn D, Hellman R, Hirsch I, Inzucchi S, Ismail-Beigi F, Kirkman M, Umpierrez G; American Association of Clinical Endocrinologists; American Diabetes Association. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care. 2009;32(6):1119-31
83. Hu F, Stampfer M, Haffner S, Solomon C, Willett W, Manson J. Elevated risk of cardiovascular disease prior to clinical diagnosis of type 2 diabetes. Diabetes Care. 2002;25(7):1129-34
84. Haffner S, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339(4):229-34
85. Laakso M. Cardiovascular disease in type 2 diabetes: challenge for treatment and prevention. J Intern Med. 2001;249(3):225-35
86. Hill D, Fisher M. The effect of intensive glycaemic control on cardiovascular outcomes. Diabetes Obes Metab. 2010;12(8):641-7
87. Nathan D, Cleary P, Backlund J, Genuth S, Lachin J, Orchard T, Raskin P, Zinman B; Diabetes Control, Complications Trial/Epidemiology of Diabetes Interventions, Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353(25):2643-53
88. Brown A, Reynolds L, Bruemmer D. Intensive glycemic control and cardiovascular disease: an update. Nat Rev Cardiol. 2010;7(7):369-75
89. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352(9131):837-53
90. Holman R, Paul S, Bethel M, Matthews D, Neil H. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359(15):1577-89
91. Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein H, Miller M, Byington R, Goff D Jr, Bigger J, Buse J, Cushman W, Genuth S, Ismail-Beigi F, Grimm R Jr, Probstfield J, Simons-Morton D, Friedewald W. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358(24):2545-59
92. ADVANCE Collaborative Group, Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, Hamet P, Harrap S, Heller S, Liu L, Mancia G, Mogensen C, Pan C, Poulter N, Rodgers A, Williams B, Bompoint S, de Galan B, Joshi R, Travert F. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358(24):2560-72
93. Skyler J, Bergenstal R, Bonow R, Buse J, Deedwania P, Gale E, Howard B, Kirkman M, Kosiborod M, Reaven P, Sherwin R; American Diabetes Association; American College of Cardiology Foundation; American Heart Association. Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE, and VA diabetes trials: a position statement of the American Diabetes Association and a scientific statement of the American College of Cardiology Foundation and the American Heart Association. Diabetes Care. 2009;32(1):187-92
94. Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven P, Zieve F, Marks J, Davis S, Hayward R, Warren S, Goldman S, McCarren M, Vitek M, Henderson W, Huang G; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360(2):129-39
95. Duckworth W. VA Diabetes Trial (VADT) Update. 69th Scientific Sessions, American Diabetes Association. 2009
96. American Diabetes Association. Standards of medical care in diabetes-2011. Diabetes Care. 2011;34(Suppl 1):S11-61
97. Chalmers J, Cooper M. UKPDS and the legacy effect. N Engl J Med. 2008;359(15):1618-20
98. Control Group, Turnbull F, Abraira C, Anderson R, Byington R, Chalmers J, Duckworth W, Evans G, Gerstein H, Holman R, Moritz T, Neal B, Ninomiya T, Patel A, Paul S, Travert F, Woodward M. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia. 2009;52(11):2288 -
99. ACCORD Study Group, Buse J, Bigger J, Byington R, Cooper L, Cushman W, Friedewald W, Genuth S, Gerstein H, Ginsberg H, Goff D Jr, Grimm R Jr, Margolis K, Probstfield J, Simons-Morton D, Sullivan M. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am Cardiol. 2007;99(12A):21i-33i
100. Gerstein H, Riddle M, Kendall D, Cohen R, Goland R, Feinglos M, Kirk J, Hamilton B, Ismail-Beigi F, Feeney P; ACCORD Study Group. Glycemia treatment strategies in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol. 2007;99(12A):34i-43i
101. Polonsky W, Fisher L, Schikman C, Hinnen D, Parkin C, Jelsovsky Z, Amstutz L, Schweitzer M, Wagner R. The value of episodic, intensive blood glucose monitoring in non-insulin treated persons with Type 2 Diabetes: design of the Structured Testing Program (STeP) study, a cluster-randomised, clinical trial [NCT00674986]. BMC Fam Pract. 2010;11:37
102. Polonsky W, Fisher L, Schikman C, Hinnen D, Parkin C, Jelsovsky Z, Petersen B, Schweitzer M, Wagner RS. Structured self-monitoring of blood glucose significantly reduces A1C levels in poorly controlled, noninsulin- treated type 2 diabetes: results from the Structured Testing Program study. Diabetes Care. 2011;34(2):262-7
103. Polonsky W, Jelsovsky Z, Panzera S, Parkin C, Wagner R. Primary care physicians identify and act upon glycemic abnormalities found in structured, episodic blood glucose monitoring data from non-insulin-treated type 2 diabetes. Diabetes Technol Ther. 2009;11(5):283-91
104. Polonsky W, Fisher L, Schikman C, Hinnen D, Parkin C, Jelsovsky Z, Schweitzer M, Petersen B, Wagner R. A structured self-monitoring of blood glucose approach in type 2 diabetes encourages more frequent, intensive, and effective physician interventions: results from the STeP study. Diabetes Technol Ther. 2011;13(8):797-802
105. Polonsky W, Fisher L, Schikman C, Hinnen D, Parkin C, Jelsovsky Z, Petersen P, Schweitzer M, Wagner R. Structured blood glucose monitoring intervention leads to significant glycemic improvement in poorly controlled, non-insulin treated type 2 diabetes: Results from the STeP Study. Diabetologia. 2010;53(Suppl 1):S417
106. Karter A, Parker M, Moffet H, Spence M, Chan J, Ettner S, Selby J. Longitudinal study of new and prevalent use of self-monitoring of blood glucose. Diabetes Care. 2006;29(8):1757-63
107. Guerci B, Drouin P, Grangé V, Bougnères P, Fontaine P, Kerlan V, Passa P, Thivolet C, Vialettes B, Charbonnel B; ASIA Group. Self-monitoring of blood glucose significantly improves metabolic control in patients with type 2 diabetes mellitus: the Auto-Surveillance Intervention Active (ASIA) study. Diabetes Metab. 2003;29(6):587-94
108. Schwedes U, Siebolds M, Mertes G; SMBG Study Group. Meal-related structured self-monitoring of blood glucose: effect on diabetes control in non- insulin-treated type 2 diabetic patients. Diabetes Care. 2002;25(11):1928-32
109. Barnett A, Krentz A, Strojek K, Sieradzki J, Azizi F, Embong M, Imamoglu S, Perusicová J, Uliciansky V, Winkler G. The efficacy of self-monitoring of blood glucose in the management of patients with typ 2 diabetes treated with a gliclazide modified release-based regimen. A multicentre, randomized, parallel-group, 6-month evaluation (DINAMIC 1 study). Diabetes Obes Metab. 2008;10(12):1239-47
110. Durán A, Martín P, Runkle I, Pérez N, Abad R, Fernández M, Del Valle L, Sanz M, Calle-Pascual A. Benefits of self-monitoring blood glucose in the management of new-onset Type 2 diabetes mellitus: the St Carlos Study, a prospective randomized clinic-based interventional study with parallel groups. J Diabetes. 2010;2(3):203-11
111. Bonomo K, De Salve A, Fiora E, Mularoni E, Massucco P, Poy P, Pomero A, Cavalot F, Anfossi G, Trovati M. Evaluation of a simple policy for pre- and post-prandial blood glucose self-monitoring in people with type 2 diabetes not on insulin. Diabetes Res Clin Pract. 2010;87(2):246-51
112. Farmer A, Wade A, Goyder E, Yudkin P, French D, Craven A, Holman R, Kinmonth A, Neil A. Impact of self monitoring of blood glucose in the management of patients with non-insulin treated diabetes: open parallel group randomised trial. BMJ. 2007;335(7611):132
113. Davidson M, Castellanos M, Kain D, Duran P. The effect of self monitoring of blood glucose concentrations on glycated hemoglobin levels in diabetic patients not taking insulin: a blinded, randomized trial. Am J Med. 2005;118(4):422-5
114. O'Kane M, Bunting B, Copeland M, Coates V; ESMON study group. Efficacy of self monitoring of blood glucose in patients with newly diagnosed type 2 diabetes (ESMON study): randomised controlled trial. BMJ. 2008;336(7654):1174-7
115. Nalysnyk L, Hernandez-Medina M, Krishnarajah G. Glycaemic variability and complications in patients with diabetes mellitus: evidence from a systematic review of the literature. Diabetes Obes Metab. 2010Apr;12(4):288-98
Author contact
![]() Corresponding author: Rolf Hinzmann, M.D., Ph.D., Roche Diagnostics GmbH, Diabetes Care, Sandhofer Straße 116, 68305 Mannheim / Germany. Telephone: +49 621 759 2254; Fax: +49 621 759 5831; rolf.hinzmanncom.
Corresponding author: Rolf Hinzmann, M.D., Ph.D., Roche Diagnostics GmbH, Diabetes Care, Sandhofer Straße 116, 68305 Mannheim / Germany. Telephone: +49 621 759 2254; Fax: +49 621 759 5831; rolf.hinzmanncom.