Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2012; 9(7):555-566. doi:10.7150/ijms.4455 This issue Cite
Research Paper
Cytotoxicity of 15-Deoxy-Δ12,14-prostaglandin J2 through PPARγ-independent Pathway and the Involvement of the JNK and Akt Pathway in Renal Cell Carcinoma
Megumi Fujita1, Chiaki Tohji1, Yoko Honda1, Yasuhiro Yamamoto2, Tsutomu Nakamura2, Tatsurou Yagami2, Motohiro Yamamori1, Noboru Okamura1 ![]()
1. Department of Clinical Pharmacy, School of Pharmacy and Pharmaceutical Sciences, Mukogawa Women's University, 11-68 Koshien-kyuban-cho, Nishinomiya, Hyogo 663-8179, Japan;
2. Department of Pharmaceutical Health Care, Faculty of Pharmaceutical Sciences, Himeji Dokkyo University, 2-1 Kami-ohno 7-Chome, Himeji, Hyogo 670-8524, Japan.
Received 2012-4-9; Accepted 2012-8-31; Published 2012-9-4
Abstract
Introduction: Agonists of peroxisome proliferator-activated receptor gamma (PPARγ) have been examined as chemopreventive and chemotherapeutic agents. The aim was to investigate the cytotoxicity and action mechanisms of 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), one of endogenous ligands for PPARγ, in terms of PPARγ-dependency and the mitogen-activated protein kinase (MAPK) and Akt pathway in three human renal cell carcinoma (RCC)-derived cell lines.
Methods: 786-O, Caki-2 and ACHN cells were used as human RCC-derived cell lines. Cell viability and caspase-3 activity was detected by fluorescent reagents, and chromatin-condensation was observed with a brightfield fluorescent microscope after staining cells with Hoechst33342. The expression levels of proteins were detected by Western blot analysis.
Results: 15d-PGJ2 showed cytotoxicity in dose-dependent manner. 15d-PGJ2 induced chromatin-condensation and elevated caspase-3 activity, and the cell viability was restored by co-treatment with a pan-caspase inhibitor, Z-VAD-FMK, indicating the involvement of caspase-dependent apoptosis. The cytotoxicity was not impaired by a PPARγ inhibitor, GW9662, suggesting that 15d-PGJ2 exerted the cytotoxicity in a PPARγ-independent manner. Some antioxidants rescued cells from cell death induced by 15d-PGJ2, but some did not, suggesting that reactive oxygen species (ROS) did not contribute to the apoptosis. 15d-PGJ2 also increased the expression levels of phospho-c-Jun N terminal kinase (JNK) in Caki-2 cells, and decreased those of phospho-Akt in 786-O cells, indicating that the JNK MAPK and the Akt pathways participated in the anticancer effects of 15d-PGJ2 in some cell lines.
Conclusion: 15d-PGJ2 exerted cytotoxic effects accompanying caspase-dependent apoptosis, and this effect was elicited in a PPARγ-independent manner in three cell lines. In addition, the JNK MAPK and Akt pathway was involved in the cytotoxicity of 15d-PGJ2 to some extent in some cell line. Therefore, our study showed the 15d-PGJ2 to potentially be an interesting approach for RCC treatment.
Keywords: 15-deoxy-Δ12,14prostaglandin J2, PPARγ, Renal cell carcinoma, Apoptosis, JNK MAPK, Akt.
Introduction
Renal cell carcinoma (RCC) accounts for about 2% of all tumors, and the most common histological subtype is clear renal cell carcinoma (75%) [1]. Although nephrectomy is the most effective treatment at an early stage, advanced renal cancer is still associated with a poor prognosis, with a five-year survival rate of less than 10%, as RCC is relatively resistant to chemotherapy and radiotherapy [2]. Recently, novel agents for RCC targeting cancer-specific pathways have been developed, such as sorafenib and sunitinib. Although they have been shown to be beneficial in patients with advanced RCC [3, 4], the effect is insufficient and it is therefore necessary to discover new targets for the treatment of RCC. Indeed, there have been several reports of other possible targets by us and others [4-6].
Peroxisome proliferator-activated receptor gamma (PPARγ) belongs to the superfamily of nuclear hormone receptor transcription factors [7], playing major roles in adipogenesis [8], glucose metabolism [9], and angiogenesis [10]. It has been reported that PPARγ is expressed in normal tissues and its expression is changed at a variety of tumor sites [11]. It has been also detected in the tissue of renal cell carcinoma patients at the mRNA and protein level [12, 13].
15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) is an end-product of prostaglandin D2, and known as an endogenous ligand for PPARγ [14, 15]. Badawi et al. showed that tissue levels of 15d-PGJ2 were markedly decreased in tumors and metastatic breast tissue compared with control tissue, suggesting that the modulation in tissue levels of 15d-PGJ2 influence the development of cancer and its progression to metastasis [16]. Therefore, it is thought to be well worth investigating the effects of 15d-PGJ2 on cancer cells as a potential treatment for cancer. Indeed, 15d-PGJ2 shows not only anti-inflammatory and cytoprotective activities, but also proapoptotic and anti-proliferative properties in many cancer cell lines [11], including colorectal [17-20], lung [20], breast [20, 21], hepatocellular [22], renal cell [23-25], prostate, and bladder cancers [25, 26]. These apoptotic effects are thought to be mediated by PPARγ-dependent [27, 28] and PPARγ-independent [19, 21, 22, 26, 29] pathways. Moreover, a number of apoptotic mechanisms have been proposed for 15d-PGJ2 [11, 14, 21, 27, 30-33], for example, the involvement of the mitogen-activated protein kinase (MAPK) pathway [17, 18, 29, 31]; however, little information on RCC has been available to date. Furthermore, we already demonstrated that 15d-PGJ2 enhanced the anti-tumor activity of camptothecin against Caki-2 cells, one of renal carcinoma cell lines independently of PPARγ pathway [34].
In the present study, we investigated whether 15d-PGJ2 induced cell death in three human RCC cell lines, 786-O (Von Hippel-Lindau (VHL) -deficient and primary organ-derived cells), Caki-2 (VHL-expressing and primary organ-derived cells) and ACHN (VHL-expressing and metastatic site-derived cells). We chose them because all three cell lines have been commonly used and we could elucidate whether they would respond to 15d-PGJ2 through a similar mechanism or not. We also examined its mechanisms of action in terms of dependency on PPARγ and the MAPK and Akt pathways, comparing among three cell lines.
Materials and methods
Chemicals
15d-PGJ2 was purchased from Cayman Chemical (Ann Arbor, MI) and dissolved in DMSO just before use. The final concentration of DMSO in the medium did not exceed 0.1%. GW9662 was purchased from Sigma (St. Louis, MO). N-benzyloxycarbonyl-Val-Ala-Asp fluoromethyl ketone (Z-VAD-FMK) was obtained from Enzo Life Sciences International (Plymouth Meeting, PA). N-acetyl-L-cysteine (NAC), reduced glutathione (GSH), vitamin E, and melatonin were purchased from Nacalai Tesque (Kyoto, Japan). SB202190 (p38 inhibitor), SP600125 (c-Jun N-terminal kinase (JNK) inhibitor), and Akt inhibitor IV were obtained from Merck (Darmstadt, Germany).
Cells and cell culture
786-O, ACHN, and Caki-2 cells were obtained from Summit Pharmaceuticals International (Tokyo, Japan), the Cell Resource Center for Biomedical Research Institute of Development, Aging and Cancer Tohoku University (Sendai, Japan), and DS Pharma Biomedical (Osaka, Japan), respectively, and were used as human renal carcinoma cell models. Then 786-O and Caki-2 were maintained in a culture medium consisting of RPMI1640 (Nacalai Tesque) and ACHN was cultured in DMEM (Wako Pure Chemical Industries, Osaka, Japan). The media were supplemented with 10% heat-inactivated FBS (Invitrogen, Life Technologies, Carlsbad, CA) and 50 U/mL penicillin-50 µg/mL streptomycin (Nacalai Tesque). Cells were cultured in an atmosphere of 95% air and 5% CO2 at 37˚C, and subcultured every 3 or 4 days.
Cell viability
Cell viability was measured with Cell Quanti-BlueTM (BioAssay Systems, Hayward, CA). Briefly, cells were seeded onto 96-well plates (Asahi Glass, Tokyo, Japan) at 5 × 103 /well and incubated for 24 h. The cells were treated with 15d-PGJ2 or Akt inhibitor IV in the presence or absence of other chemicals for a further 24 h using FBS-free medium. The assay was performed as described previously [6]. It utilizes the conversion of alamar blue reagent to resorufin fluorescence by metabolically active cells and resorufin was measured in a CytoFluor® Series 4000 Fluorescence Multi-Well Plate Reader (PerSeptive Biosystems, Framingham, MA) at an excitation wavelength of 530 nm / emission 580 nm. The 50% growth inhibitory concentrations (IC50) were calculated according to the sigmoid inhibitory effect model E = IC50γ/(IC50γ + Cγ). E represents the surviving fraction (% of control), and C and γ represent the drug concentration in the medium and the Hill coefficient, respectively. For co-exposure experiments, 15d-PGJ2 was used at 1.5, 7 and 3 μM for 786-O, Caki-2, and ACHN cell lines, respectively, as determined approximately the IC50 of each cell line. The agents and their concentrations used for co-exposure experiments are indicated as follow: Z-VAD-FMK (100 µM), GW9662 (20 µM), NAC (1 mM), GSH (1 mM), vitamin E (1 µM), melatonin (1 mM), SB202190 (3 µM), and SP600125 (1 µM for 786-O and Caki-2 cells / 0.1, 0.3, and 1 µM for ACHN cells).
Detection of chromatin condensation (fluorescence microscopy)
For nuclear staining, cells were treated with 15d-PGJ2 for 24 h at 1.5, 7 and 3 μM for 786-O, Caki-2 and ACHN cell lines, respectively. Immediately after, the nuclear chromatin of trypsinized cells was stained with 80 μg/mL of Hoechst 33342 (Nacalai Tesque) in the dark at room temperature for 15 min. They were then observed with a brightfield fluorescent microscope (VANOX; Olympus, Tokyo, Japan) under UV excitation. Chromatin-condensed cells were photographed at a 40-fold magnification. In addition, at a 20-fold magnification, more than 120 cells with condensed chromatin were counted in each experiment and their percentage was calculated.
Fluorimetric assay of caspase-3 activity
Caspase-3 activity was assessed using a Caspase 3 Assay Kit, Fluorimetric (Sigma), according to the manufacturer's instructions. Briefly, cells were seeded in 96-well plates at 5 × 103 /well and cultured for 24 h. After exposure to 15d-PGJ2 at 1.5, 7 and 3 μM for 786-O, Caki-2, and ACHN cell lines, respectively, the supernatant was aspirated and cells were harvested with lysis buffer (50 mM HEPES, pH 7.4, 5 mM CHAPS and 5 mM DTT). The reaction buffer, containing 16.6 μM Acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (Ac-DEVD-AMC), a caspase-3-specific substrate, was added to the wells, and the production of AMC was detected in a CytoFluor® Plate Reader at 360 nm (excitation) / 460 nm (emission). Enzymatic activity was determined as initial velocity expressed as nmol AMC/min/mL. It was then corrected with the quantity of protein in each well detected by a BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA).
Assay of lactate dehydrogenase (LDH) leakage
Cells were seeded onto 24-well plates (Asahi glass) at 3 × 104 /well and cultured for 24 h. The medium was removed and the cells were washed with fresh serum-free medium 3 times and incubated for 1 h. They were treated with 15d-PGJ2 at 1.5, 7 and 3 μM for 786-O, Caki-2, and ACHN cell lines, respectively, for 24 h, and supernatant was obtained by centrifugation (170 × g, 3 min). The LDH activity in the supernatant was assayed with LDH-Cytotoxic Test Wako (Wako), according to the manufacturer's instructions. Briefly, the samples were reacted with a coloring solution containing nitrotetrazolium blue, and the diformazan produced from nitrotetrazolium blue by oxidation of NADH was detected in a MTP-500 microplate reader (CORONA ELECTRIC, Ibaraki, Japan) at 570 nm. LDH leakage was calculated according to the equation; LDH leakage (% per positive control) = (S-N) / (P-N). S, N and P represent the absorbance in the sample, negative and positive controls, respectively. The positive control was the supernatant of cells treated with 0.2% Tween 20 (Nacalai Tesque), and the negative control was the supernatant incubated with 0.1% DMSO.
Antibodies
The rabbit monoclonal antibodies against phospho-SAPK/JNK (Thr183/Tyr185), Akt (C67E7) and phospho-Akt (Ser 473) (D9E) XP™ were purchased from Cell Signaling Technology (Danvers, MA) and the mouse monoclonal antibody against β-actin (C4), from Santa Cruz Biotechnology (Santa Cruz, CA). The horseradish peroxidase-linked antibody for donkey anti-rabbit IgG and sheep anti-mouse IgG was obtained from GE Healthcare (Buckinghamshire, UK). All antibodies were diluted with blocking buffer.
Western blot analysis
ACHN cells were harvested by scraping and washed with ice-cold phosphate-buffered saline (PBS). In the experiment for phosphorylation of JNK MAPK, cells were plated in 100-mm dishes 24 h before treatment and then treated with 15d-PGJ2 at 3 μM for 1, 8 or 24 h, and in the experiment for phosphorylation of Akt, cells were treated with 15d-PGJ2 for 1 and 24 h. Lysis buffer (20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 1 mM EDTA, 0.1% SDS, 1mM NaF, 1 mmol/L Na3VO4 and 0.1% protease inhibitor cocktail (Sigma)) was added to pellets, and the cells were sonicated briefly then incubated on ice for 20 min. Cell extracts were centrifuged at 16,000 × g for 15 min at 4˚C, and the supernatants were transferred to new tubes. Protein concentrations were determined by a BCA protein assay kit (Thermo Fisher Scientific). The samples were mixed with the same volume of Laemmli Sample buffer containing β-mercaptoethanol (Bio-Rad Laboratories, Hercules, CA) and boiled for 5 min, and 15 μg of protein was loaded onto 10% SDS-polyacrylamide gels. After electrophoresis, the proteins were transferred to polyvinylidene difluoride membranes (ATTO, Tokyo, Japan) and blocked with Tris-buffered saline-0.1% Tween 20 (TBS-T) containing 2% ECL Advance™ Blocking Agent (GE Healthcare) for 1 h. Blocked membranes were reacted with primary antibodies (diluted 1:10,000) for 1 h at room temperature and washed five times with TBS-T. After incubation with the secondary antibody (diluted 1:25,000) for 1 h at room temperature, the membranes were again washed five times. The signal was visualized using ECL Advance™ detection reagents (GE Healthcare). Protein expression levels were evaluated on densitometric measurements using ImageJ software. β-actin level was used as a standard for each sample, and protein levels at base line (0 h) were defined as 1.0.
Statistical analysis
Data are expressed as the mean ± SD. Statistical significance was compared with Student's or Welch's t-test for comparisons of two groups and non-repeated one-way ANOVA followed by Dunnett's post-hoc test for multiple comparisons. P < 0.05 was considered significant.
Results
Effects of 15d-PGJ2 on cell viability
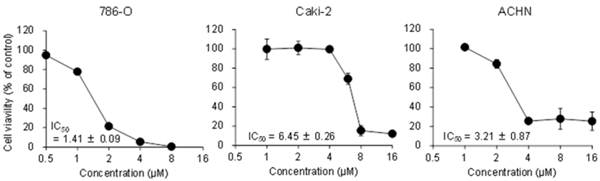
Figure 1 shows the effects of 15d-PGJ2 on cell viability in three RCC cell lines. 15d-PGJ2 showed cytotoxicity in a dose-dependent manner with IC50 values of 1.41 ± 0.09, 6.45 ± 0.26, and 3.21 ± 0.87 µM in 786-O, Caki-2, and ACHN cells, respectively, suggesting that 15d-PGJ2 showed cytotoxicity in RCC cell lines.
Concentration-dependent cytotoxic effects of 15d-PGJ2 in 786-O, Caki-2, and ACHN cells. Cells were precultured for 24 h at 5 × 103 /well in 96-well plates and treated with 15d-PGJ2 for 24 h. Cell viability was assessed by fluorescent assay, and data represent the mean ± S.D. from 4 independent preparations.
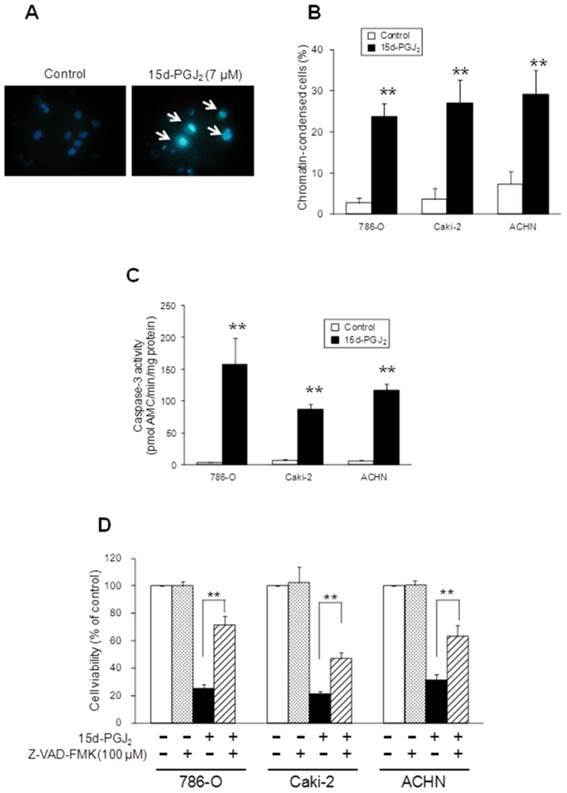
Effects of 15d-PGJ2 on nuclear morphology and caspase-3 activity
To explore whether the cytotoxicity of 15d-PGJ2 occurred through apoptosis or necrosis, we observed chromatin condensation, a typical morphological change in apoptosis, and the activation of caspase-3, a downstream factor of the caspase cascade. When cells were exposed to 15d-PGJ2 at approximately the IC50 of each cell line for 24 h, chromatin condensation was found after staining with Hoechst 33342 (Figure 2A). The percentage of cells with chromatin condensation was significantly increased by 15d-PGJ2 treatment, from 3 to 24, 4 to 27 and 7 to 30% in 786-O, Caki-2, and ACHN cells, respectively (Figure 2B). Next, we examined the activation of caspase-3. Treatment with 15d-PGJ2 resulted in approximately a 36-, 12-, and 19-fold increase of caspase activity in 786-O, Caki-2, and ACHN cells, respectively (Figure 2C). Moreover, a pan-caspase inhibitor, Z-VAD-FMK, significantly but not completely recovered the viability of cells exposed to 15d-PGJ2, from 25 to 71%, 21 to 47%, and 31 to 63%, respectively (Figure 2D). These results suggested that 15d-PGJ2 induced cell death through caspase-dependent apoptosis to significant extent.
Apoptosis assays for 15d-PGJ2 in three RCC cell lines. (A, B) Induction of chromatin condensation by 15d-PGJ2. Typical fluorescence micrographs of Caki-2 cells stained with Hoechst 33342 (A) and percentages of chromatin-condensed cells (B). Cells were treated with 15d-PGJ2 for 24 h at the IC50 of each cell line and stained with Hoechst 33342 for 15 min at room temperature. They were then observed under a brightfield fluorescence microscope with UV excitation and the percentage of chromatin-condensed cells was determined. Data represent the mean ± S.D. from 4 independent preparations. Statistically significant difference from the control: **p < 0.01 assessed by t-test. (C) Effects of 15d-PGJ2 on caspase-3 activity. Cells were seeded in 96-well plates at 5 × 103 /well and cultured for 24 h. After exposure to 15d-PGJ2 at the IC50 of each cell line for 24 h, caspase-3 activity was assessed with a fluorimetric Caspase 3 Assay Kit according to the manufacturer's instructions. Enzymatic activities were determined as initial velocities corrected with the quantity of protein. Data represent the mean ± S.D. from 4 independent preparations. Statistically significant difference from the control: **p < 0.01 assessed by t-test. (D) Effects of a pan-caspase inhibitor on 15d-PGJ2-induced cell death. Cells were precultured for 24 h at 5 × 103 /well in 96-well plates and exposed to 15d-PGJ2 with or without Z-VAD-FMK (100 μM) for 24 h. Cell viability was assessed by fluorescent assay, and data represent the mean ± S.D. from 4 to 5 independent preparations. Statistical significance was assessed by t-test.
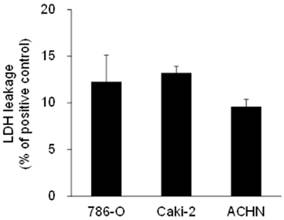
Effects of 15d-PGJ2 on LDH leakage
To find the contribution of necrosis to cell death, we next measured the levels of LDH in media as a marker of cell lysis and necrosis [35]. LDH leakage per positive control were 12.3 ± 2.9%, 13.2 ± 0.7%, and 9.6 ± 0.8% in 786-O, Caki-2, and ACHN cells, respectively, when cells were treated with approximately the IC50 of 15d-PGJ2 (Figure 3), suggesting that necrosis was partly involved in cell death.
LDH leakage in RCC cell lines treated with 15d-PGJ2. Cells were precultured for 24 h at 3 × 104 /well in 24-well plates and exposed to 15d-PGJ2 for 24 h. Absorbance at 570 nm in supernatant of the sample (S), negative control (N), and positive control (P) was used for calculation. LDH leakage was determined at follows; LDH leakage (% per positive control) = (S-N) / (P-N), and data represent the mean + S.D. from 3 independent preparations. The positive control was the supernatant of cells treated with 0.2% Tween 20 and the negative control was the supernatant of cells incubated with 0.1% DMSO.
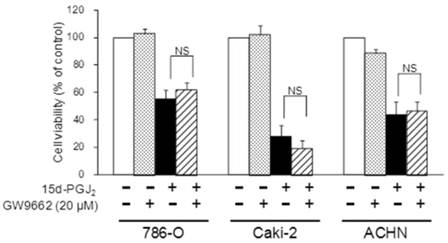
Participation of PPARγ in the effect of 15d-PGJ2 on cell viability
To investigate the molecular mechanisms of cell death induced by 15d-PGJ2, we first examined PPARγ-dependency. Figure 4 shows the effects of a PPARγ antagonist, GW9662, on 15d-PGJ2-induced cell death. The cytotoxic effect of 15d-PGJ2 was not impaired by GW9662, indicating it to be independent of PPARγ.
Effects of a PPARγ inhibitor, GW9662, on 15d-PGJ2-induced cell death. Cells were precultured for 24 h at 5 × 103 /well in 96-well plates and exposed to 15d-PGJ2 with or without GW9662 (20 μM) for 24 h. Cell viability was assessed by fluorescent assay, and data represent the mean ± S.D. from 5 independent preparations. Statistical significance was assessed by t-test.
The effects of antioxidants on the cytotoxicity of 15d-PGJ2
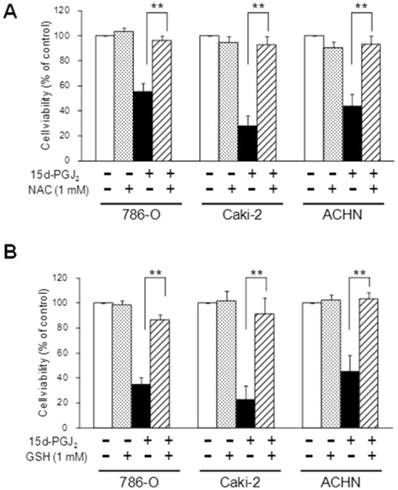
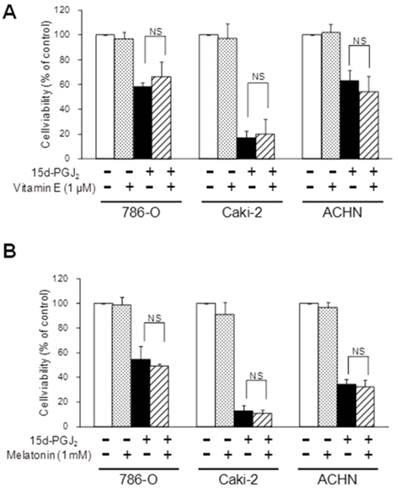
As 15d-PGJ2 was also reported to generate reactive oxygen species (ROS) [36], we next investigated the effects of antioxidants. Figure 5 shows the effect of antioxidants, NAC and GSH, on the cytotoxic action of 15d-PGJ2. NAC or GSH almost completely reversed the 15d-PGJ2-induced cell death in all three cell lines. Conversely, there was no recovery at all from 15d-PGJ2-induced cell death among cells co-treated with other antioxidants, vitamin E (1 µM) and melatonin (1 mM) (Figure 6).
Effects of antioxidants with a thiol-group, NAC (A) and GSH (B), on 15d-PGJ2-induced cell death. Cells were precultured for 24 h at 5 × 103 /well in 96-well plates and exposed to 15d-PGJ2 in the presence or absence of NAC (500 μM) or GSH (1 mM) for 24 h. Cell viability was assessed by fluorescent assay, and data represent the mean ± S.D. from 4 to 5 independent preparations. Statistical significance was assessed by t-test.
Effects of antioxidants without a thiol-group, vitamin E (A) and melatonin (B), on 15d-PGJ2-induced cell death. Cells were precultured for 24 h at 5× 103 /well in 96-well plates and exposed to 15d-PGJ2 in the presence or absence of vitamin E (1 μM) or melatonin (1 mM) for 24 h. Cell viability was assessed by fluorescent assay, and data represent the mean ± S.D. from 4 to 5 independent preparations. Statistical significance was assessed by t-test.
Participation of the p38 and JNK MAPK pathways in the effect of 15d-PGJ2 on cell viability
To investigate the involvement of the MAPK pathway in the cytotoxic mechanisms of 15d-PGJ2, two MAPK inhibitors were co-treated with 15d-PGJ2. Figure 7A shows the effects of a p38 inhibitor, SB202190, on 15d-PGJ2-induced cell death. Cell viability was not impaired by co-exposure to SB202190, suggesting that the p38 pathway did not mainly participate in the cytotoxic effect of 15d-PGJ2.
Involvement of the p38 and JNK MAPK pathway in 15d-PGJ2-induced cell death. Effects of a p38 MAPK inhibitor, SB202190 (A), or JNK inhibitor, SP600125 (B, C), on 15d-PGJ2-induced cell death and the effects of 15d-PGJ2 on phosphorylation of JNK (D). Cells were precultured for 24 h at 5 × 103 /well in 96-well plates and exposed to 15d-PGJ2 at approximately the IC50 in the presence or absence of SB202190 (3 μM) or SP600125 (0.1, 0.3, 1 μM) for 24 h. Cell viability was assessed by fluorescent assay, and data represent the mean ± S.D. from 4 independent preparations. Statistical significance was assessed by t-test or Dunnett's test. To detect proteins, cells were precultured for 24 h in 100-mm dishes. Cells were then treated with 15d-PGJ2 at 3 μM for 0 and 8 h. Protein (15 μg) was analyzed by Western blotting for the expression of phospho-JNK. Relative protein levels were quantified using ImageJ, and each phospho-JNK signal was normalized to the β-actin signal. The representative bands and the results of densitometric analysis from three independent preparations were described, and data represent the mean ± S.D. from 4 independent preparations.
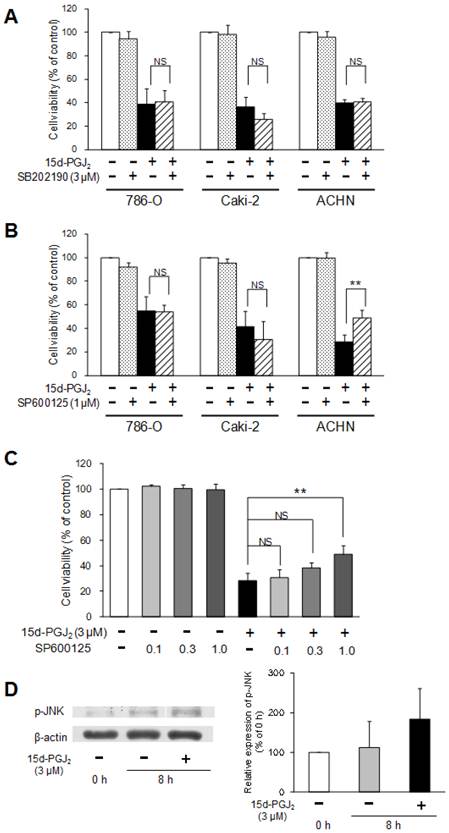
A JNK inhibitor, SP600125, did not prevent 15d-PGJ2-induced cell death in 786-O and Caki-2 cells, but significantly improved the viability of ACHN cells (Figure 7B).
As improvement of cell viability by SP600125 (1 µM) was observed only in ACHN cells, we next investigated the dose-dependent effects of SP600125 and the effects of 15d-PGJ2 on the expression of phospho-JNK in ACHN cells. SP600125 improved the viability of ACHN cells in a dose-dependent manner (Figure 7C). In addition, the expression levels of phospho-JNK was tend to be increased by exposure to 15d-PGJ2 for 8 h (Figure 7D). This suggested the JNK pathway to be involved in 15d-PGJ2-induced cell death to some extent in ACHN cells.
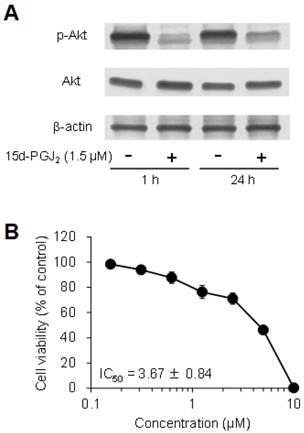
Participation of the Akt pathways in the effect of 15d-PGJ2 on cell viability
The expression levels of phospho-Akt were markedly decreased by exposure to 15d-PGJ2 for 1 and 24 h only in 786-O cells, whereas the levels of total Akt were not changed (Figure 8A), but not in other cells (data not shown). In addition, Akt inhibitor IV decreased the cell viability of 786-O cells in a dose-dependent manner, with an IC50 value of 3.67 ± 0.84 μM (Figure 8B). This suggested that 15d-PGJ2 inhibited Akt activation at the same levels of an Akt inhibitor and exerted cytotoxic effects in 786-O cells.
Involvement of the Akt pathway in 15d-PGJ2-induced cell death. (A) To detect proteins, cells were precultured for 24 h in 100-mm dishes. Cells were then treated with 15d-PGJ2 at 3 μM for 1 and 24 h. Protein (15 μg) was analyzed by Western blotting for the expression of Akt and phospho-Akt. (B) To investigate the effects of an Akt inhibitor, cells were precultured for 24 h at 5 × 103 /well in 96-well plates and treated with Akt inhibitor IV for 24 h. Cell viability was assessed by fluorescent assay, and data represent the mean ± S.D. from 6 independent preparations. Relative protein levels were quantified using ImageJ, and each Akt and phospho-Akt signal was normalized to the β-actin signal. Relative ratio of normalized Akt and phospho-Akt signal to untreated cells in each time was described.
Discussion
PPARγ is known to play many roles when activated by a ligand, such as synthetic antidiabetic thiazolidinediones and a natural eicosanoid derivate, 15d-PGJ2. These ligands, apart from their functions in normal tissues, have been reported to induce growth arrest or apoptosis in diverse tumor cells [6, 11, 13, 17-27, 29, 30]. In the present study, we evaluated the effects of 15d-PGJ2 on the viability of RCC cells and investigated the mechanisms of cytotoxicity.
First, we confirmed the cytotoxicity of 15d-PGJ2 in RCC cell lines as it was demonstrated in Caki-2 cells in the previous study [34]. 15d-PGJ2 exhibited dose-dependent cytotoxicity with IC50 values of 1.4-6.5 μM (Figure 1). This cytotoxicity of 15d-PGJ2 accompanied chromatin condensation (Figure 2A, 2B) and caspase-3 activation (Figure 2C). In addition, a pan-caspase inhibitor, Z-VAD-FMK, significantly restored cell viability (Figure 2D), suggesting that 15d-PGJ2 induced cell death through caspase-dependent apoptosis to some extent in RCC cell lines; however, the recovery achieved by Z-VAD-FMK was incomplete. This suggested some involvement of other forms of death, such as caspase-independent apoptosis, autophagy, or necrosis. Indeed, a caspase-independent mechanism was reported to be partly involved in the 15d-PGJ2-induced apoptosis of hepatoma, chondrosarcoma, and choriocarcinoma cells [22, 37, 38]. Moreover, LDH leakage was observed in all three cell lines treated with 15d-PGJ2, albeit to a small extent (Figure 3), suggesting that necrosis was partly but not mainly involved in the cell death.
To investigate the molecular mechanisms of 15d-PGJ2-induced cell death, we first examined the involvement of PPARγ. 15d-PGJ2 has high affinity for human PPARγ [15], and is known to induce adipogenesis through PPARγ-activation, but it is unclear whether PPARγ is involved in the cytotoxic effect of 15d-PGJ2. In our results, GW9662, an irreversible PPARγ antagonist, did not improve cell viability at all (Figure 4), although the PPARγ protein is expressed in these three cell lines [6]. This suggested the cytotoxicity of 15d-PGJ2 in RCC cell lines to be independent of PPARγ, as supported by reports in other cell types [19, 21, 22, 26, 29]. Kim et al. showed that GW9662 inhibited the cytotoxic effect of 15d-PGJ2 in neuroblastoma cells [27]. Shen et al. showed that cell death was partly, but not completely, blocked by GW9662 in chondrosarcoma cells, suggesting that 15d-PGJ2 exerted its effect via both PPARγ-dependent and -independent pathways [30]. Accordingly, the involvement of PPARγ in the cytotoxicity of 15d-PGJ2 seems to depend on the cell strain, and it is important to accumulate information for each type of cell as the genomic responses to PPARγ activation are complex [11].
15d-PGJ2 was also reported to generate ROS and to induce apoptosis in various kinds of cells [17, 18, 28, 36], so we next investigated the effects of antioxidants. Interestingly, our data showed that two antioxidants, NAC and GSH, rescued cells from cell death induced by 15d-PGJ2 (Figure 5), although the other antioxidants, vitamin E and melatonin, did not (Figure 6). These findings suggested that NAC and GSH recovered the cell viability by the mechanisms other than the antioxidative effects, and this discrepancy among the antioxidants might be accounted for by their construction. As 15d-PGJ2 can reportedly form covalent adducts with thiol-containing biomolecules via Michael addition [14], due to an electrophilic α,β-unsaturated carbonyl group in the cyclopentenone ring, NAC and GSH, possessing a thiol-group, were assumed to bind to 15d-PGJ2 and attenuate the effect of 15d-PGJ2. In addition, Paumi et al. showed that a GSH-conjugate of 15d-PGJ2 was effluxed from cells through multidrug resistance proteins, and the cytotoxic and transactivating effects of 15d-PGJ2 were attenuated in breast cancer cells [39]. Accordingly, in this case as well, NAC, a precursor of GSH, was converted to GSH, and could reduce the intracellular concentration of 15d-PGJ2, thereby recovering the cell viability. Therefore, these findings indicated that ROS did not contribute to the apoptosis induced by 15d-PGJ2 in RCC cells.
Next, we focused on the cell signaling pathways involved in cell survival or proliferation. As some reports showed that 15d-PGJ2 also regulated MAPK activation [17, 18, 29, 31], we first investigated the involvement of the p38 and JNK MAPK pathways, known to be activated by various forms of stress, in the cytotoxic effects of 15d-PGJ2 on RCC cells. While a p38 MAPK inhibitor (SB202190) did not affect the cytotoxicity of 15d-PGJ2 in any of the cell lines (Figure 7A), a JNK inhibitor (SP600125) improved the viability of 15d-PGJ2-treated ACHN cells in a dose-dependent manner (Figure 7B, 7C), and the level of phosphorylated JNK tended to be increased after 8 h (Figure 7D). These findings suggested that the p38 pathway did not mainly participate in the 15d-PGJ2-elicited cell death, but that the JNK pathway was involved to some extent only in ACHN cells. The reason why the mechanisms of action differed among the three cell lines was unclear; but Okano et al. also reported that the actions of 15d-PGJ2 differed even among different cell lines derived from the organ [22]. In addition, ACHN cells are derived from a metastatic site (pleural effusion), while 786-O and Caki-2 cells originate from a primary organ, and this might partly affect the cell-dependency.
We next investigated the involvement of Akt, which is known to be a downstream of phosphatidylinositol 3-kinase (PI3K) and have potent antiapoptotic and proliferative functions. 15d-PGJ2 markedly decreased the expression level of phosphorylated Akt after 1 and 24 h-exposure in 786-O cells (Figure 8A). This suggested that 15d-PGJ2 inactivated Akt and exerted cytotoxicity in 786-O cells, in which an Akt inhibitor also showed cytotoxicity with a low IC50 value (Figure 8B). The detail mechanisms of inactivating Akt must be further investigated, but, like other reports, the up-regulation of PTEN followed by the reduction of PI3K activity might be involved in this case [40]. Therefore, the inactivation of Akt observed in this study might be an important mechanism of 15d-PGJ2 in some cell lines like 786-O cells.
In conclusion, the cytotoxicity of 15d-PGJ2 were mainly due to caspase apoptosis in three RCC cell lines but through different mechanisms in terms of JNK MAPK and Akt pathways. Our study showed the treatment with 15d-PGJ2 to potentially be an interesting approach for RCC though further experimental and clinical investigations are needed.
Acknowledgements
This work was supported by a Grant-in-aid for Scientific Research from the Ministry of Education, Culture Sports, Science, and Technology of Japan.
Authors' contributions
MF, YY, TN TY and NO made conception, designed and coordinated the study. MF and CT carried out all experiments. MF, YH and MY carried out data analysis. MF and NO prepared the manuscript. All authors read and approved the final manuscript.
Abbreviations
15d-PGJ2: 15-deoxy-Δ12,14-prostaglandin J2; AMC: 7-amido-4-methylcoumarin; GSH: Reduced glutathione; LDH: Lactate dehydrogenase; MAPK: Mitogen-activated protein kinase; NAC: N-acetyl-L-cysteine; PPARγ: Peroxisome proliferator-activated receptor gamma; RCC: Renal cell carcinoma; ROS: Generate reactive oxygen species; TBS: Tris-buffered saline; VHL: von Hippel-Lindau; Z-VAD-FMK: N-benzyloxycarbonyl-Val-Ala-Asp fluoromethyl ketone.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. McLaughlin JK, Lipworth L, Tarone RE. Epidemiologic aspects of renal cell carcinoma. Semin Oncol. 2006;33:527-533
2. Amato RJ. Renal cell carcinoma. Review of novel single agent therapeutics and combination regimens. Ann Oncol. 2005;16:7-15
3. Pirrotta MT, Bernardeschi P, Fiorentini G. Targeted-therapy in advanced renal cell carcinoma. Curr Med Chem. 2011;18:1651-1657
4. Flaherty KT, Puzanov I. Building on a foundation of VEGF and mTOR targeted agents in renal cell carcinoma. Biochem Pharmacol. 2010;80:638-646
5. Cho DC, Cohen MB, Panka DJ, Collins M, Ghebremichael M, Atkins MB, Signoretti S, Mier JW. The efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 compared with rapamycin in renal cell carcinoma. Clin Cancer Res. 2010;16:3628-3638
6. Fujita M, Yagami T, Fujio M, Tohji C, Takase K, Yamamoto Y, Sawada K, Yamamori M, Okamura N. Cytotoxicity of troglitazone through PPARγ-independent pathway and p38 MAPK pathway in renal cell carcinoma. Cancer Lett. 2011;312:219-227
7. Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645-650
8. Camp HS, Ren D, Leff T. Adipogenesis and fat-cell function in obesity and diabetes. Trends Mol Med. 2002;8:442-447
9. Rangwala SM, Lazar MA. Peroxisome proliferator-activated receptor γ in diabetes and metabolism. Trends Pharmacol Sci. 2004;25:331-336
10. Fauconnet S, Lascombe I, Chabannes E, Adessi GL, Desvergne B, Wahli W, Bittard H. Differential regulation of vascular endothelial growth factor expression by peroxisome proliferator-activated receptors in bladder cancer cells. J Biol Chem. 2002;277:23534-23543
11. Koeffler HP. Peroxisome proliferator-activated receptor γ and cancers. Clin Cancer Res. 2003;9:1-9
12. York M, Abdelrahim M, Chintharlapall S, Lucero SD, Safe S. 1,1-Bis(3'-Indolyl)-1-(p-Substitutedphenyl)methanes induce apoptosis and inhibit renal cell carcinoma growth. Clin Cancer Res. 2007;13:6743-6752
13. von Schwarzenberg K, Held SA, Schaub A, Brauer KM, Bringmann A, Brossart P. Proteasome inhibition overcomes the resistance of renal cell carcinoma cells against the PPARγ ligand troglitazone. Cell Mol Life Sci. 2009;66:1295-1308
14. Kim EH, Surh YJ. 15-Deoxy-Δ12,14-prostaglandin J2 as a potential endogenous regulator of redox-sensitive transcription factors. Biochem Pharmacol. 2006;72:1516-1528
15. Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83:813-819
16. Badawi AF, Badr MZ. Expression of cyclooxygenase-2 and peroxisome proliferator-activated receptor-γ and levels of prostaglandin E2 and 15-deoxy-Δ12,14-prostaglandin J2 in human breast cancer and metastasis. Int J Cancer. 2003;103:84-90
17. Grau R, Iñiguez MA, Fresno M. Inhibition of activator protein 1 activation, vascular endothelial growth factor, and cyclooxygenase-2 expression by 15-deoxy-Δ12,14-prostaglandin J2 in colon carcinoma cells: evidence for a redox-sensitive peroxisome proliferator-activated receptor-γ-independent mechanism. Cancer Res. 2004;64:5162-5171
18. Shin SW, Seo CY, Han H, Han JY, Jeong JS, Kwak JY, Park JI. 15d-PGJ2 induces apoptosis by reactive oxygen species-mediated inactivation of Akt in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clin Cancer Res. 2009;15:5414-5425
19. Schaefer KL Takahashi H, Morales VM Harris G, Barton S Osawa E, Nakajima A Saubermann LJ. PPARγ inhibitors reduce tubulin protein levels by a PPARγ, PPARδ and proteasome-independent mechanism, resulting in cell cycle arrest, apoptosis and reduced metastasis of colorectal carcinoma cells. Int J Cancer. 2006;120:702-713
20. Allred CD, Kilgore MW. Selective activation of PPARγ in breast, colon, and lung cancer cell lines. Mol Cell Endocrinol. 2005;235:21-29
21. Clay CE, Monjazeb A, Thorburn J, Chilton FH, High KP. 15-Deoxy-Δ12,14-prostaglandin J2-induced apoptosis does not require PPARγ in breast cancer cells. J Lipid Res. 2002;43:1818-1828
22. Okano H, Shiraki K, Inoue H, Yamanaka Y, Kawakita T, Saitou Y, Yamaguchi Y, Enokimura N, Yamamoto N, Sugimoto K, Murata K, Nakano T. 15-deoxy-Δ-12-14-PGJ2 regulates apoptosis induction and nuclear factor-κB activation via a peroxisome proliferator-activated receptor-γ-independent mechanism in hepatocellular carcinoma. Lab Invest. 2003;83:1529-1539
23. Yuan J, Takahashi A, Masumori N, Itoh N, Tsukamoto T. Peroxisome proliferator-activated receptor gamma is frequently underexpressed in renal cell carcinoma. Int J Urol. 2006;13:265-270
24. Yuan J, Takahashi A, Masumori N, Uchida K, Hisasue S, Kitamura H, Itoh N, Tsukamoto T. Ligand for peroxisome proliferator-activated receptor gamma have potent antitumor effect against human renal cell carcinoma. Urology. 2005;65:594-599
25. Yoshimura R, Matsuyama M, Hase T, Tsuchida K, Kuratsukuri K, Kawahito Y, Sano H, Segawa Y, Nakatani T. The effect of peroxisome proliferator-activated receptor-γ ligand on urological cancer cells. Int J Mol Med. 2003;12:861-865
26. Chaffer CL, Thomas DM, Thompson EW, Williams ED. PPARγ-independent induction of growth arrest and apoptosis in prostate and bladder carcinoma. BMC Cancer. 2006;6:53
27. Kim EJ, Park KS, Chung SY, Sheen YY, Moon DC, Song YS, Kim KS, Song S, Yun YP, Lee MK, Oh KW, Yoon DY, Hong JT. Peroxisome proliferator-activated receptor-γ activator 15-deoxy-Δ12,14-prostaglandin J2 inhibits neuroblastoma cell growth through induction of apoptosis: association with extracellular signal-regulated kinase signal pathway. J Pharmacol Exp Ther. 2003;307:505-517
28. Li L, Tao J, Davaille J, Feral C, Mallat A, Rieusset J, Vidal H, Lotersztajn S. 15-deoxy-Δ12,14-prostaglandin J2 induces apoptosis of human hepatic myofibroblasts. A pathway involving oxidative stress independently of peroxisome-proliferator-activated receptors. J Biol Chem. 2001;276:38152-38158
29. Lennon AM, Ramaugé M, Dessouroux A, Pierre M. MAP kinase cascades are activated in astrocytes and preadipocytes by 15-deoxy-Δ12-14-prostaglandin J2 and the thiazolidinedione ciglitazone through peroxisome proliferator activator receptor γ-independent mechanisms involving reactive oxygenated species. J Biol Chem. 2002;277:29681-29685
30. Shen ZN, Nishida K, Doi H, Oohashi T, Hirohata S, Ozaki T, Yoshida A, Ninomiya Y, Inoue H. Suppression of chondrosarcoma cells by 15-deoxy-Δ12,14-prostaglandin J2 is associated with altered expression of Bax/Bcl-xL and p21. Biochem Biophys Res Commun. 2005;328:375-382
31. Ho TC, Chen SL, Yang YC, Chen CY, Feng FP, Hsieh JW, Cheng HC, Tsao YP. 15-deoxy-Δ12,14-prostaglandin J2 induces vascular endothelial cell apoptosis through the sequential activation of MAPKS and p53. J Biol Chem. 2008;283:30273-30288
32. Kondo M, Shibata T, Kumagai T, Osawa T, Shibata N, Kobayashi M, Sasaki S, Iwata M, Noguchi N, Uchida K. 15-Deoxy-Δ12,14-prostaglandin J2: the endogenous electrophile that induces neuronal apoptosis. Proc Natl Acad Sci U S A. 2002;99:7367-7372
33. Kang DS, Kwon CH, Park JY, Kim JH, Woo JS, Jung JS, Kim YK. 15-deoxy-Δ12,14-prostaglandin J2 induces renal epithelial cell death through NF-κB-dependent and MAPK-independent mechanism. Toxicol Appl Pharmacol. 2006;216:426-435
34. Yamamoto Y, Fujita M, Koma H, Yamamori M, Nakamura T, Okamura N, Yagami T. 15-deoxy-Δ12,14-prostaglandin J2 enhanced the anti-tumor activity of camptothecin against renal cell carcinoma independently of topoisomerase-II and PPARγ pathways. Biochem Biophys Res Commun. 2011;410:563-567
35. Kim YM, Chung HT, Simmons RL, Billiar TR. Cellular non-heme iron content is a determinant of nitric oxide-mediated apoptosis, necrosis, and caspase inhibition. J Biol Chem. 2000;275:10954-10961
36. Atarod EB, Kehrer JP. Dissociation of oxidant production by peroxisome proliferator-activated receptor ligands from cell death in human cell lines. Free Radic Biol Med. 2004;37:36-47
37. Nishida K, Furumatsu T, Takada I, Kawai A, Yoshida A, Kunisada T, Inoue H. Inhibition of human chondrosarcoma cell growth via apoptosis by peroxisome proliferator-activated receptor-γ. Br J Cancer. 2002;86:1303-1309
38. Keelan JA, Sato TA, Marvin KW, Lander J, Gilmour RS, Mitchell MD. 15-Deoxy-Δ12,14-prostaglandin J2, a ligand for peroxisome proliferator-activated receptor-γ, induces apoptosis in JEG3 choriocarcinoma cells. Biochem Biophys Res Commun. 1999;262:579-585
39. Paumi CM, Wright M, Townsend AJ, Morrow CS. Multidrug resistance protein (MRP) 1 and MRP3 attenuate cytotoxic and transactivating effects of the cyclopentenone prostaglandin, 15-deoxy-Δ12,14prostaglandin J2 in MCF7 breast cancer cells. Biochemistry. 2003;42:5429-5437
40. Na HK, Surh YJ. Peroxisome proliferator-activated receptor γ (PPARγ) ligands as bifunctional regulators of cell proliferation. Biochem Pharmacol. 2003;66:1381-1391
Author contact
![]() Corresponding author: Noboru Okamura, PhD., Department of Clinical Pharmacy, School of Pharmacy and Pharmaceutical Sciences, Mukogawa Women's University, 11-68 Koshien-kyuban-cho, Nishinomiya, Hyogo 663-8179, Japan. Tel/Fax: +81 798 45 9955, E-mail: nokamuraac.jp.
Corresponding author: Noboru Okamura, PhD., Department of Clinical Pharmacy, School of Pharmacy and Pharmaceutical Sciences, Mukogawa Women's University, 11-68 Koshien-kyuban-cho, Nishinomiya, Hyogo 663-8179, Japan. Tel/Fax: +81 798 45 9955, E-mail: nokamuraac.jp.