Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2012; 9(5):370-379. doi:10.7150/ijms.4395 This issue Cite
Research Paper
18α-Glycyrrhetinic Acid Down-Regulates Expression of Type I and III Collagen via TGF-Β1/Smad Signaling Pathway in Human and Rat Hepatic Stellate Cells
Lei Zong1,2, Ying Qu1, Ming-yi Xu1, Yu-wei Dong1, Lun-gen Lu1 ![]()
1. Department of Gastroenterology, Shanghai First People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200080, China
2. Department of Gastroenterology, Weifang People's Hospital, Weifang, Shandong Province 261041, China
Received 2012-3-22; Accepted 2012-6-18; Published 2012-7-10
Abstract
Objective: To investigate the effects of 18α-glycyrrhetinic acid (18α-GA) on the expression of type I and III collagen in human and rat hepatic stellate cells (HSC) and to explore the role of TGF-β1/Smad signaling pathway involved.
Methods: Following 18α-GA treatment, the cell viability and cell growth were detected to determine the optimal concentration of 18α-GA. The expressions of TGF-β1/Smad signaling-related genes including type I and III collagen in human and rat HSCs before and after 18α-GA treatment were measured by real time PCR. The expression of related proteins was verified by western blot assay. The phosphorylation level of Smad2 and Smad3 was detected by immunocytochemistry. The DNA binding activities of SP-1, AP-1 and NF-κB were measured by both EMSA and ArrayStar transcription factor activity assay.
Results: 18α-GA could decrease the mRNA and protein expression of Smad3, type I and III collagen, increase the Smad7 expression in human and rat HSCs (P<0.05), and reduce phosphorylation level of Smad3 at 24 h and 48 h after treatment. The DNA binding activities of transcription factors were suppressed by 18α-GA in human and rat HSCs at 24 h, and the activities reduced in a time dependent manner with the lowest activities at 48 h, especially for SP-1.
Conclusion: 18α-GA could inhibit the mRNA and protein expression of type I and III collagen in human and rat HSCs, which may be attributed to down-regulation of Smad3, up-regulation of Smad7, and inhibition of DNA binding activities of SP-1, AP-1 and NF-κB.
Keywords: 18α-glycyrrhetinic acid, hepatic stellate cell, TGF-β1/Smad, transcription factor
Introduction
Hepatic fibrosis occurs as a wound-healing process in different chronic liver injuries, including hepatic viral infection, autoimmune liver diseases and alcoholic liver disease. Hepatic fibrogenesis is a process in which the production of extracellular matrix (ECM) surpasses its degradation. In the absence of effective treatments, reversible hepatic fibrosis at an early stage may become irreversible resulting in cirrhosis. Hepatic cirrhosis is often accompanied by hepatic failure due to impaired liver function and associated with portal hypertension due to hemodynamic alterations. Thus, to reverse the hepatic fibrogenesis and prevent the occurrence of cirrhosis have attracted increasing attention from physicians on hepatology in clinical practice.
The cells primarily responsible for ECM production during the hepatic fibrosis are hepatic stellate cells (HSCs). Upon stimulation, quiescent HSCs become activated and transdifferentiates into myofibroblast-like cells characterized by several key phenotypic changes, including an increase in proliferation, accumulation of ECM, expression of α-smooth muscle actin (α-SMA) and loss of stored vitamin A droplets. Growth factors that are particularly important for this progress include transforming growth factor β (TGF-β), which is essential for HSCs transdifferentiation in vivo [1]. TGF-β binding to the type II TGF-β receptor (TβRII) leads to the recruitment, phosphorylation, and activation of the type I TGF-β receptor (TβRI). The activated TβRI kinase then phosphorylates Smad2 or Smad3, and Smad2/3 complex recruits Smad4 to form a Smad2/3/4 complex, which finally enters the nucleus activating the target genes such as type I collagen, type III collagen and fibronectin [2-6].
The most important bioactive compounds of licorice root (Glycyrrhiza radix) are the triterpene glycoside glycyrrhizic acid (glycyrrhizin, GL) and its aglycone 18β-glycyrrhetinic acid (GA). 18β-GA, one of the most important bioactive compounds in licorice root, has two configuration epimers on 18H, α and β body. 18β-GA is a major component of natural licorice, and can be transformed into 18α-GA. Both compounds are reported to have anti-tumor, anti-inflammatory and antiviral properties. It decreases the serum level of alanine aminotransferase (ALT) in patients with chronic hepatitis C (CHC) and suppresses the progression of hepatic fibrosis as well as subsequent occurrence of hepatocellular carcinoma (HCC) [7]. Although there were several previous studies on 18β-GA, the specific mechanisms of bioeffects of 18α-GA are virtually unknown. The present study aimed to investigate the effect of 18α-GA on expression of type I and III collagen in cultured HSCs and to explore the role of TGF-β1/Smad signaling pathway in this effect.
Materials and methods
Cell lines, culture, and treatment
The human stellate cell line (LX-2 cells) and rat stellate cell line (CCl4-cirrhotic fat-storing cells, CFSC) were kindly provided by the Prof Friedman (Mt. Sinai Medical Center, NY, USA) and were the spontaneously immortalized cells [8]. They exhibit typical features of activated stellate cells: expression of desmin and glial acidic fibrillary protein (GAFP), and responsiveness to platelet-derived growth factor (PDGF) BB and TGF-β. These cells express α-smooth muscle actin (α-SMA) under all culture conditions, and thus can be regarded as at least partially activated, even after immediate replating. Cells were incubated for 24 h and 48 h in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) (Gibco Products International Incorporated, Los Angeles, CA, USA) and 2 mg/ml of 18α-GA (Sigma-Aldrich Co., St. Louis, MO, USA) in the absence of antibiotics in an incubator at 37oC with 5% CO2. 18α-GA was added to the DMEM at final concentration of 8 μM.
Determination of cytotoxicity of LX-2 cells to 18α-GA
LX-2 cells were seeded into 96-well plates (5 000/well; Corning, USA) and incubated in DMEM containing 10% FCS for 4 h. When cells became adherent, 18α-GA was added to each well at final concentrations of 0~40 mg/ml followed by incubation for 48 h. Then, the medium was refreshed (100 μl), and cell counting kit-8 (CCK-8) solution was added (10 μl/well) followed by incubation for 1.5 h. Absorbance was measured at 450 nm on a Model 680 Microplate Reader (Bio-Rad, USA.). The half maximal inhibitory concentration (IC50) was calculated according to the manufacturer's instructions (Dojindo Laboratories, Japan). Six wells were included in each group and experiment was done at least thrice.
Detection of cell growth
For detection of cell growth, cells were seeded into 24-well plates (2×104/well), and incubated in DMEM containing 10% FCS overnight. After incubation in serum free medium for 24 h, cells were maintained in DMEM containing 0.4% FCS or subsequently stimulated with 10% FCS in the presence or absence of 18α-GA. The final concentration of 18α-GA was 8 μM. At pre-designed time points, cell count was determined with a computer-equipped cell counter (Nucleocounter, ChemoMetec Corporation, USA.). Each treatment was done in triplicate and experiment was performed at least thrice.
RNA extraction and real time PCR
RNA extraction was carried out by using Trizol (Invitrogen Co., Carlsbad, CA, USA) and real time PCR performed according to the manufacturer's instructions using M-MLV reverse transcriptase (Takara Shuzo Co., Ltd, Japan) and real time PCR Master Mix (SYBR Green) Kit (Toyobo Co., Ltd. Japan). GAPDH served as an internal control. The primers are listed in Table 1 and 2. The PCR conditions were 95°C for 10 min, and 40 cycles of 95°C for 20 s, 54°C for 30 s and 72°C for 30s. Each experiment was performed thrice in triplicate. The fold-change in mRNA of target gene relative to that of GAPDH was calculated according to previously described [9].
Primers for target genes in LX-2
| Genes | Primers | Size (bp) |
|---|---|---|
| Smad2 | F: 5'- GCCATCACCACTCAAAACTGT -3' R:5'- GCCTGTTGTATCCCACTGATCTA-3 | 111 |
| Smad3 | F: 5'- ATCCTGCCTTTCACTCCCC -3' R: 5'- CTGCCCCGTCTTCTTGAGTT -3' | 132 |
| Smad4 | F: 5'- ACGAACGAGTTGTATCACCTGG -3' R: 5'- ATGGCTGTCCCTCAAAGTCAT -3' | 114 |
| Smad7 | F: 5'-GGCCGGATCTCAGGCATTC-3' R: 5'- GAGTCGGCTAAGGTGATGGG-3' | 110 |
| TβRⅠ | F:5'-TGGCGGGGAGAAGAAGTTG-3' R:5'-CTGCTGCTATAAATCCCAGGATG-3' | 127 |
| TβRⅡ | F:5'-AGAAGTCGGATGTGGAAATGGA-3' R: 5'-CTGCACCGTTGTTGTCAGTG-3' | 123 |
| COL1A2 | F:5'-AATTGGAGCTGTTGGTAACGC-3' R: 5'- CACCAGTAAGGCCGTTTGC -3' | 125 |
| COL3A1 | F:5'-TGGTCCCCAAGGTGTCAAAG-3' R:5'-GGGGGTCCTGGGTTACCATTA-3' | 117 |
| GAPDH | F:5'-CATGAGAAGTATGACAACAGCCT-3' R:5'- AGTCCTTCCACGATACCAAAGT -3' | 113 |
primers for target genes in CFSC
| Genes | Primers | Size (bp) |
|---|---|---|
| Smad2 | F: 5'- GATGGTCGTCTTCAGGTGTCTC -3' R: 5'- CCTCTGGTAGTGGTAAGGGTTC -3' | 174 |
| Smad3 | F: 5'-AAATGACAGCAGCAGGGACACTA -3' R: 5'-TGAGGAGGTAGGACCCACAGTAGA-3' | 176 |
| Smad4 | F: 5'- ATCACTATGAGCGGGTTGTC -3' R: 5'- GAATGTCCTTCCGTGGGTAA -3' | 142 |
| Smad7 | F: 5'- CTCGGAAGTCAAGAGGCTGTG -3' R: 5'- CCATCGGGTATCTGGAGTAAGGA -3' | 137 |
| TβRⅠ | F: 5'- CTGCCTGCTTCTCATCGTGT -3' R: 5'- AACTTTGTCTGTGGTCTCGG -3' | 154 |
| TβRⅡ | F: 5'- CCC TACTCTGTCTGTGGATGA -3' R: 5'- GACGTCATTTCCCAGAGTAC -3' | 152 |
| COL1A2 | F: 5'- TGACCAGCCTCGCTCACAG-3' R: 5'- 5 GCGGGCAGGGTTCTTTCTA -3' | 127 |
| COL3A1 | F: 5'- TCCCAGAACATTACATACCACT -3' R: 5'- GCTATTTCCTTCAGCCTTGA -3' | 126 |
| GAPDH | F: 5'- GATGAGATTGGCATGGCTTT -3' R: 5'- GAGAAGTGGGGTGGCTT -3' | 196 |
Western Blot assay
Proteins were extracted from cells and Western blot assay was performed according to the standard protocol. Treated cells were harvested at appropriate time points and mixed in ice-cold lysis buffer containing 1% PMSF. After centrifugation, the supernatant was collected and then subjected to SDS polyacrylamide gel electrophoresis. The proteins were then transferred to a PVDF membrane which was subsequently blocked for 1 h at room temperature in Tris-buffered saline containing 0.1% Tween 20 and 3% BSA. Then, the membrane was incubated with following antibodies overnight at 4°C: anti-TβRI, anti-TβRII, anti-Smad2, anti-Smad3, anti-Smad4 anti-Smad7, anti-COLI, anti-COLIII (Abcam, Cambridgeshire, England). GAPDH (Beyotime, Jiangsu Province, China) served as an internal control. The concentration of primary antibody was obtained from the manufacturer's instructions. Detection was done with enhanced chemiluminescence (Invitrogen, Carlsbad, CA, USA). Bands were scanned and analyzed using NIH Image J (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD). Experiment was performed in triplicate for at least three times.
Immunocytochemistry
LX-2 and CFSC were seeded onto coverslip in plates. When cells were adherent, cells were treated with 8 μM 18α-GA for 24 h and 48 h and then fixed in 4% formaldehyde for 15 min followed by incubation with primary antibodies (anti-phosphorylation of Smad2 [Abcam, Cambridgeshire, England] and anti-phosphorylation of Smad 3 [Novus Biologicals,Colorado, USA]) for 1 h and subsequently with horseradish peroxidase conjugated secondary antibodies (Maixin company, Fuzhou, China). DAB kit (Maixin company, Fuzhou, China) was used for visualization for no more than 3 min.
Electrophoreticmobilityshiftassay (EMSA)
Nuclear extracts were isolated from LX-2 and CFSC using the NucBuster Protein Extraction kit (Beyotime, Jiangsu Province, China) according to the manufacturer's instructions. The biotin-conjugated double-stranded oligonucleotides were used including commercially available specificity protein-1 (SP-1), activator protein (AP-1), nuclear factor-κB (NF-κB). The sequences of probes are listed in Table 3 (Beyotime, Jiangsu Province, China). Specific binding was confirmed by competition experiments with a 100-fold excess of unlabeled, identical oligonucleotides. After binding, the samples were separated by non-denaturating PAGE and bands were detected by NIH Image J (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD, USA).
Sequence of probes
| TF | Probes | |
|---|---|---|
| NF-κB | Probe | 5´-AGTTGAGGGGACTTTCCCAGGC-3´ 3'-TCA ACTCCCCTGAAAGGGTCCG-5' |
| Mutant | 5´-AGTTGAGGCGACTTTCCCAGGC -3´ 3'-TCA ACTCCGCTGAAAGGGTCCG-5' | |
| AP-1 | Probe | 5'-CGCTTGATGACTCAGCCGGAA-3' 3'-GCGAACTACTGAGTCGGCCTT-5' |
| Mutant | 5'-CGCTTGATGACTTGGCCGGAA-3' 3'-GCGAACTACTGAACCGGCCTT-5' | |
| SP-1 | Probe | 5'-ATTCGATCGGGGCGGGGCGAGC-3' 3'-TAAGCTAGCCCCGCCCCGCTCG-5' |
| Mutant | 5'-ATTCGATCGGTTCGGGGCGAGC-3' 3'-TAAGCTAGCCAAGCCCCGCTCG-5' | |
Detection of DNA-binding activity
Nuclear extracts were prepared as described previously. The multiwell colorimetric assay for phosphorylated p65 protein was performed using the Trans-AM NF-κB p65 Transcription Factor Assay Kit (Shanghai KangChen Co., China). Briefly, nuclear extracts of equal amount were incubated in 96-well plates coated with immobilized oligonucleotides which included SP-1, AP-1 and NF-κB binding site. Transcription factors binding to target oligonucleotides were detected by incubation with primary antibodies and horseradish peroxidase conjugated secondary antibody. For quantification of activity, optical densities (OD) were measured at 450 nm with a Model 680 Microplate Reader (Bio-Rad Laboratories, Berkeley, California, USA).
Statistical analysis
Data were presented as mean ± standard deviation (S.D.). Differences between groups were evaluated using an unpaired two-sided Student's t-test. Comparisons of multiple groups with control group were done with ANOVA followed by Dunnett's test for post hoc analysis. A value of P<0.05 was considered statistically significant.
Results
18α-GA inhibited LX-2 cell growth in a dose-dependent manner
Cells were incubated for 48 h after treatment with 18α-GA, and viable cells counted. The cell viability was above 80% at concentrations up to 16 μM. The number of apoptotic LX-2 after treatment with 18α-GA at concentrations up to 8 μM was comparable to that in the control group. Thus, 8 μM 18α-GA was used in the following experiments.
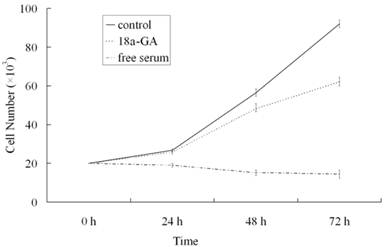
To further investigate the effect of 18α-GA on LX-2 cell growth, serum-free medium treated cells were maintained in DMEM containing 0.4% FCS or stimulated with 10% FCS in the presence or absence of 18α-GA at indicated concentrations for 24, 48 or 72 h and cell growth was measured. As shown in Fig. 1, at 24 h after treatment with 18α-GA at 8 μM, LX-2 cell growth was markedly reduced (P<0.05). At 48 h and 72 h after treatment with 8 μM 18α-GA, cell growth was further inhibited. At 72 h, 18α-GA at 8 μM decreased cell numbers by 23.14%, 39.66%, 56.12%, 64.68%, respectively (P<0.05).
Effects of 18α-GA on LX-2 cell growth in a dose and time dependent manner.
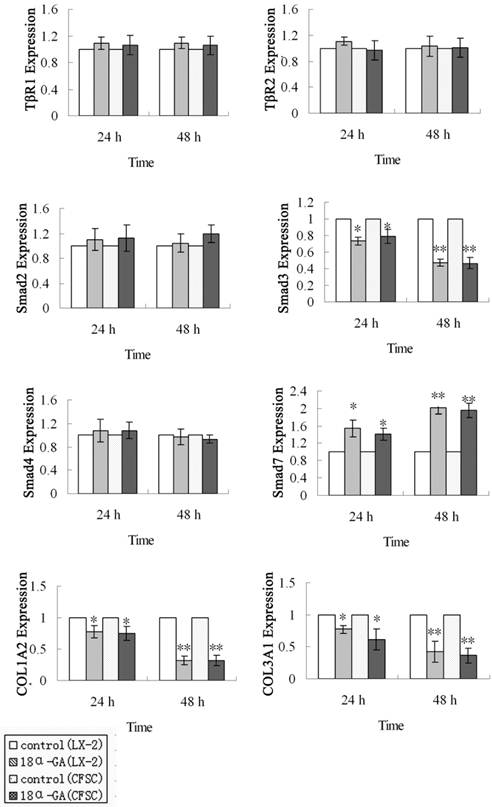
Effects of 18α-GA on mRNA expression of TGF-β1/Smad signaling pathway related proteins in human and rat HSCs
First, the mRNA expression of TGF-β1/Smad signaling pathway related protein was measured by real time PCR. The mRNA expression of Smad3, Smad7, COL1A2 and COL3A1 was markedly affected after 18α-GA treatment for 24 h and 48 h. At 24 h after 18α-GA treatment for 24 h in LX-2 cells, the mRNA expression of Smad3 (0.73), COL1A2 (0.78), and COL3A1 (0.77) was significantly decreased, while that of Smad7 (1.55) dramatically increased, when compared with the control group (P<0.05). The fold-change in mRNA expression of Smad3, COL1A2, COL3A1 and Smad7 was 0.47, 0.32, 0.43, and 2.00, respectively, at 48 h after treatment (P<0.01) (Fig 2). The same trend was also observed in CFSC. The fold-change in mRNA expression of Smad3, COL1A2, COL3A1, and Smad7 was 0.79, 0.75, 0.62, and 1.42, respectively, at 24 h after treatment (P<0.05), and 0.47, 0.32, 0.36, and 1.95, respectively, at 48 h after treatment (P<0.01) (Fig 2). However, the mRNA expression of Smad2, Smad4, TβRI, TβRII remained unchanged after 18α-GA treatment.
Effect of 18α-GA on mRNA expression of TGF-β1/Smad signaling pathway related proteins in human and rat HSCs. The mRNA expression of Smad3, COLl1A2, and COL3A1 was significantly decreased, while that of Smad7 dramatically increased, However, the mRNA expression of Smad2, Smad4, TβRI, TβRII remained unchanged after 18α-GA treatment. The mRNA expression of target genes was normalized to that of GAPDH and a value of 1.0 was assigned to the mRNA expression of target genes in the control group. *P<0.05, **P<0.01 vs control.
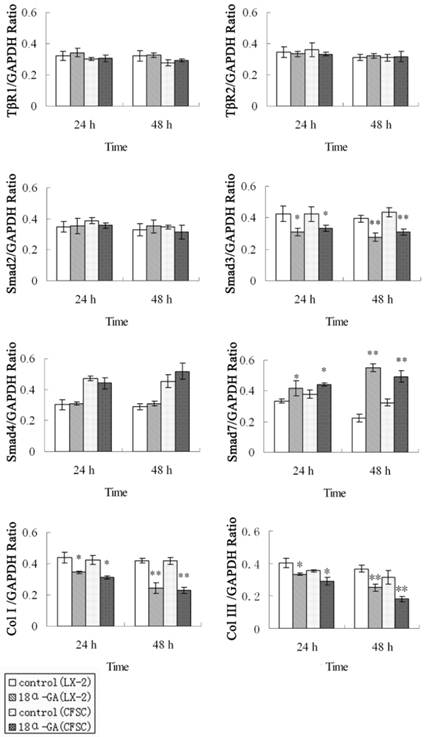
Effects of 18α-GA on the expression of TGF-β1/Smad signaling pathway related proteins in human and rat HSCs
The effect of 18α-GA on the expression of TGF-β1/Smad signaling pathway related proteins was also determined in human and rat HSCs. The changes in protein expression of Smad3, Smad7, type I and III collagen were similar to those in mRNA expression. The protein expression of Smad3, type I and III collagen was significantly decreased, while that of Smad7 dramatically increased at 24 h (P<0.05) and 48 h (P<0.01) after 18α-GA treatment in both cell lines (Fig 3).
Effect of 18α-GA on the expression of TGF-β1/Smad signaling pathway related proteins in human and rat HSCs. The protein expression of Smad3, type I and III collagen was significantly decreased, while that of Smad7 dramatically increased at 24 h (P<0.05) and 48 h (P<0.01) after 18α-GA treatment in both cell lines. * P <0.05, ** P <0.01 vs control group.
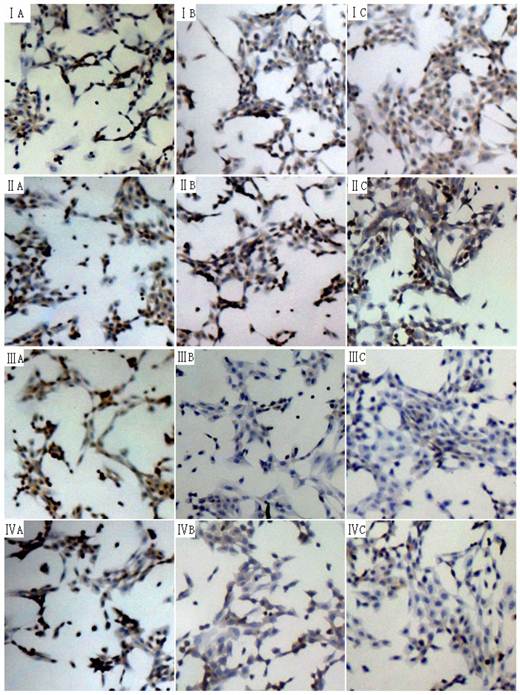
Effects of 18α-GA on the phosphorylation level of Smad2 and Smad3 in human and rat HSCs
The phosphorylation of Smad2 and Smad3 can exert important biological effects and thus the phosphorylation level of Smad2 and Smad3 in human and rat HSCs were measured by immunocytochemistry. The phosphorylation level of Smad3 was significantly reduced at 24 h and 48 h, while that of phosphorylation of Smad2 remained unchanged (Fig 4).
Effect of 18α-GA on phosphorylation level of Smad2 and Smad3 in human and rat HSCs. The phosphorylation level of Smad3 was significantly reduced at 24 h and 48 h, while that of phosphorylation of Smad2 remained unchanged. I and III, phosphorylation of Smad2; III and IV, phosphorylation of Smad3. I and III: LX-2; II and IV: CFSC. A: 0 h, B: 24 h; C: 48 h.
Effects of 18α-GA on activities of SP-1, AP-1 and NF-κB in human and rat HSCs
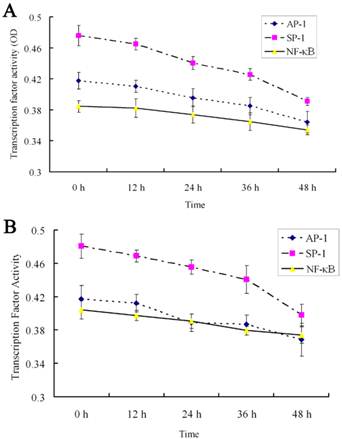
The effects of 18α-GA on the activities of SP-1, AP-1 and NF-κB in human and rat HSCs were further investigated. The activities of SP-1, AP-1 and NF-κB did not change significantly after 12h treatment with 18α-GA; the activities of SP-1, AP-1 and NF-κB decreased gradually after 24h treatment with 18α-GA which was in a time-dependent manner (Fig 5). Subsequently, the activities of above transcription factors were further detected with ArrayStar kit, and results showed similar trends in the activities of these transcription factors. There was no significant change after 12 h (P>0.05). When compared with the activities at 0 h, the inhibitory rate of SP-1 in LX-2 and CFSC was 15.7%, 22.2 %, 37.5% and 10.8%, 17.3%, 35.6% after 24h, 36h, 48h, respectively (P<0.01). The inhibitory rate of AP-1 in LX-2 and CFSC was 12.8% (P<0.05) and 18.9% (P<0.01) at 24 h, 30.7% (P<0.01) and 16.3% (P<0.05) at 36 h, and 17.2% (P<0.01) and 28.1% (P<0.01) at 48 h. The inhibitory rate of NF-κB in LX-2 and CFSC was 7.1% (P>0.05) and 13.4% (P<0.05) at 24 h, 20.4% (P<0.01) and 8.3% (P>0.05) at 36 h, and 15.3% (P<0.01) and 18.6% (P<0.01) at 48 h (Fig 5 and 6).
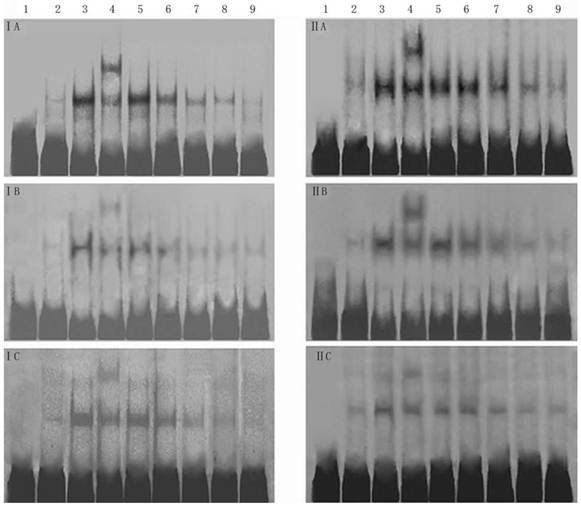
Effect of 18α-GA on DNA binding activities of transcription factors by EMSA in human and rat HSCs. The activities of SP-1, AP-1 and NF-κB decreased gradually after 18α-GA treatment which was in a time-dependent manner. I, LX-2; II , CFSC. A, AP-1; B, SP-1; C, NF-κB. 1, negative control; 2, competing reaction of probe; 3, competing reaction of mutant probe; 4, Super-shift reaction; 5-9, sample's reaction; 5, 0 h; 6, 12 h; 7, 24 h; 8, 36 h; 9, 48 h.
Effect of 18α-GA on the activities of transcription factors by ArrayStar transcription factor activity assay in human and rat HSCs. The activities of SP-1, AP-1 and NF-κB decreased gradually after 18α-GA treatment which was in a time-dependent manner. A, LX-2; B, CFSC.
Discussion
In this study, our results showed (1) 18α-GA reduced the mRNA expression of Smad3, COL1A2 and COL3A1, and increased that of Smad7 in both LX-2 and CFSC, and similar trend was also found in protein expression of these factors; (2) 18α-GA reduced the phosphorylation level of Smad3 in LX-2 and CFSC; (3) 18α-GA inhibited the activities of SP-1, AP-1 and NF-κB in both LX-2 and CFSC.
TGF-β signals are transmitted via serine / threonine kinase transmembrane receptors that phosphorylate cytoplasmic mediators of Smad family. Phospho-Smads then translocate into the nucleus, where they function as transcription factors and bind to DNA either directly or with the help of other proteins, regulating collagen I and III expression.
Over-expression of Smad3 can increase the deposition of fibronectin and collagen I and accelerate α-SMA production in HSCs [10], and HSCs from mice lacking Smad3 had decreased ability to respond to stimulation of fibers immunogenicity, indicating that Smad3 plays a promotive role in the TGF-β1-mediated fibrogenesis. Wang et al have shown that glycyrrhizin can significantly reduce the levels of Smad2 and Smad3 in primary HSCs isolated from animals with carbon tetrachloride-induced hepatic fibrosis [11]. Other studies also reveal that glycyrrhizin or glycyrrhizic acid can inhibit COL1A2 promoter activity in HSCs of mice with carbon tetrachloride-induced hepatic fibrosis, in which the decrease of Smad3 in the nucleus of HSCs plays an important role [12]. Our findings also demonstrated that the level of Smad3 was also decreased after 18α-GA treatment in a time-dependent manner in both LX-2 and CFSC, but that of Smad2 remained unchanged, which may be related to the time point and the cell type.
Ubiquitin- proteasome plays a regulatory role in the TGF-β1/Smad signaling, in which E3 ubiquitin ligase, such as Smurfs, is an important one participant. Smad7 can bind to TGF-β1 receptor complex and promote the degradation of TGF-β1 through recruiting ubiquitin ligase Smurfs. Wang et al found that the level of Smad7 was increased after administration of glycyrrhizin for 2 weeks in rats with carbon tetrachloride induced hepatic fibrosis, and the hepatic pathology presented significant improvement after 4-week treatment, suggesting that this may be due to increase of Smad7. Our results also indicated that 18α-GA increased Smad7 expression significantly in a time-dependent manner. The phosphorylation of Smad2 and Smad3 are their active form, TGF-β type I receptor and pro-inflammatory cytokine-activated kinases differentially phosphorylate Smad2 and Smad3 to create phosphoisoforms. After acute liver injury, TGF-β and pro-inflammatory cytokines synergistically enhance collagen synthesis by activated hepatic stellate cells via phosphorylation of Smad2 and Smad3 pathways [13,14]. And then the phosphorylation level of Smad2 and Smad3 was determined by immunocytochemistry in both LX-2 and CFSC. Results showed that 18α-GA reduced the phosphorylation level of Smad3, while that of phosphorylation of Smad2 remained unchanged. Previous studies have shown that Smad2 and Smad3 play different roles in the activation of HSCs: over-expression of Smad3 can significantly increase the collagen I and α-SMA expression, which is not observed after Smad2 over-expression, suggesting that Smad3 plays a more important role in the activation of HSCs [10]. Smad2 phosphorylation and nuclear translocation as a response to TGF-β1 are mainly found in resting HSCs, while Smad3 phosphorylation observed in activated HSCs. The cells in the present study were in the activated form, which may explain why no change in the phosphorylation level of Smad2 was observed.
Hepatic fibrosis is a dynamic wound healing process, during which both of the quantity and quality of ECM in the liver are significantly changed. Previous studies have revealed that the mRNA and protein expression of collagen I and III was significantly reduced in HSCs collected from rats with carbon tetrachloride and ethanol-induced hepatic fibrosis [11]. The -376~-108 bp in COL1A2 have the most potent promoter activity and contain a C/EBP binding site, three SP-1 binding sites and one AP-1 binding site. A large number of studies have shown that SP-1 and AP-1 are the transcription factors of COL1A2 as a response to TGF -β1 [15-17]. In addition, SP-1 and AP-1 can also promote the synthesis of ECM and inhibit ECM degradation through regulating TIMP, MMP and IL-6 expressions in the liver fibrogenesis [18-21].
It has been noted that NF-κB involves in the activation of HSCs [22]. Upon stimulation by cytokines, mitogen and CD40 ligand, the NF-κB activity increased rapidly, which promotes the production of intercellular adhesion molecule-1 (ICAM-1), cyclooxygenase 2 (COX2), IL-6 and IL-8 in HSCs, and triggers or exacerbates liver inflammation [23, 24].
Our findings revealed that 18α-GA could inhibit the activities of SP-1, AP-1 and NF-κB, which may also involve in the down-regulation of collagen expression after 18α-GA treatment.
However, this study was an in vitro one, and more in vivo studies are required to confirm our findings. Our results showed the expression of Smad3, Smad7, AP-1, SP-1, NF-κB, type I and III collagen was affected by 18α-GA, but we could not confirm that whether this effect was directly or indirectly related to 18α-GA. That is, the target of 18α-GA in HSCs was not determined.
In summary, our findings revealed that 18α-GA significantly reduced the expression of collagen I and III, which may be attributed to the regulation of expression of Smad3, Smad7, AP-1, SP-1 and NF-κB by 18α-GA. Furthermore, the activities of collagen-related transcription factors are also detected, and results also demonstrate that 18α-GA down-regulates the expression of type I and III collagen.
Acknowledgements
This study was supported by National Natural Science Foundation of China (No:30871162 & 81070345), the National Key Technologies Research and Development Program of China during the 11th Five-year Plan Period (2008ZX10002-006), Science and Technology Commission of Shanghai Municipality (No:09XD1403200 & 10411955300).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Derynck R, Zhang Y, Feng X-H. Smads: Transcriptional Activators of TGF-b Responses. Cell. 1998;95:737-740
2. Lagna G, Hata A, Hemmati-Brivanlou A, Massague J. Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature. 1996;383:832-836
3. Claus H, Branko S, Frank G, Elmar RB, David AB. The role of TGF-β1 in initiating hepatic stellate cell activation in vivo. J. Hepatol. 1999;30:77-87
4. Hayashi H, Abdollah S, Qiu Y. et al. The MAD-Related Protein Smad7 Associates with the TGF-β Receptor and Functions as an Antagonist of TGFβ Signaling. Cell. 1997;89:1165-1173
5. Shi YG, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685-700
6. Wu RY, Zhang Y, Feng X-H, Derynck R. Heteromeric and Homomeric Interactions Correlate with Signaling Activity and Functional Cooperativity of Smad3 and Smad4/DPC4. Mol. Cell. Biol. 1997;17:2521-2528
7. Arase Y, Ikeda K, Murashima N. et al. The long term efficacy of glycyrrhizin in chronic hepatitis C patients. Cancer. 1997;79(8):1494-1500
8. Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, Mukherjee P, Friedman SL, Eng FJ. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142-151
9. Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal. Biochem. 2000;285:194-204
10. Uemura M, Swenson ES, Gaca MDA. et al. Smad2 and Smad3 play different roles in rat hepatic stellate cell function and alpha-smooth muscle actin organization. Molecular Biology of the Cell. 2005;16(9):4214-4224
11. Wang JY, Zhang QS, Guo JS. et al. Effects of glycyrrhetinic acid on collagen Ⅰmetabolism of hepatic stellate cells at different stages of liver fibrosis in rats. World Journal of Gastroenterology. 2001;17(1):115-119
12. Moro T, Shimoyama Y, Kushida M. et al. Glycyrrhizin and its metabolite inhibit Smad3-mediated type I collagen gene transcription and suppress experimental murine liver fibrosis. Life Sci. 2008;83(15-16):531-9
13. Matsuzaki K. Smad phosphoisoform signals in acute and chronic liver injury: similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. 2012;347(1):225-243
14. Yoshida K, Matsuzaki K. Differential regulation of TGF-β/Smad signaling in hepatic stellate cells between acute and chronic liver injuries. Front Physiol. 2012;3:53
15. Inagaki Y, Kushida M, Higashi K. et al. Cell type-specific intervention of transforming growth factor β/Smad signaling suppresses collagen gene expression and hepatic fibrosis in mice. Gastroenterology. 2005;129(1):259-68
16. Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56(2):284-292
17. Zhang W, Oul J, Inagaki Y. et al. Synergistic cooperation between Sp1 and Smad3/Smad4 mediates transforming growth factor beta1 stimulation of alpha 2(I)-collagen (COL1A2) transcription. J Biol Chem. 2000;275:39237-39245
18. Takahra T, Smart DE, Oakley F. et al. Induction of myofibroblast MMP-9 transcription in three-dimensional collagen I gel cultures: regulation by NF-kappaB, AP-1 and Sp1. Int J Biochem Cell Biol. 2004;36:353-363
19. Chen A, Davis BH. NA binding protein BTEB mediates acetaldehyde-induced, jun N-terminal kinase-dependent alphaI (Ⅰ) col lagen gene expression in rat hepatic stellate cells. Mol Cell Biol. 2000;20:2818-2826
20. Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res. 2000;49:497-505
21. Kouba DJ, Chung KY, Nishiyama T. et al. Nuclear factor-kappaB mediates TNF-alpha inhibitory effect on alpha 2(I)collagen (COL1A2) gene transcription in human dermal fibroblasts. J Immunol. 1999;162:4226-4234
22. Ishida Y, Kondo T, Kimura A. et al. Absence of IL-1 receptor antagonist impaired wound healing along with aberrant NF-kappaB activation and a reciprocal suppression of TGF-beta signal pathway. J Immunol. 2006;176:5598-5606
23. Bitzer M, von Gersdorff G, Liang D. et al. A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev. 2000;14:187-197
24. Novitskiy G, Potter JJ, Rennie-Tankersley L. et al. Identification of a novel NF-kappaB-binding site with regulation of the murine alpha2(I) collagen promoter. J Biol Chem. 2004;279:15639-15644
Author contact
![]() Corresponding author: Prof. Lun-gen Lu. Department of Gastroenterology, Shanghai First People's Hospital, Shanghai Jiaotong University School of Medicine, No 100, Haining Road, Shanghai 200080 China. E-mail: lungenlu1965com. Tel: +86-21-63240090; Fax: +86-21-63241377
Corresponding author: Prof. Lun-gen Lu. Department of Gastroenterology, Shanghai First People's Hospital, Shanghai Jiaotong University School of Medicine, No 100, Haining Road, Shanghai 200080 China. E-mail: lungenlu1965com. Tel: +86-21-63240090; Fax: +86-21-63241377