Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2010; 7(3):110-119. doi:10.7150/ijms.7.110 This issue Cite
Research Paper
Parvovirus B19 Genotype Specific Amino Acid Substitution in NS1 Reduces the Protein's Cytotoxicity in Culture
Violetta Kivovich1,2, Leona Gilbert2, Matti Vuento2, Stanley J. Naides3 ![]()
1. Pennsylvania State College of Medicine/ Milton S. Hershey Medical Center, Hershey, PA, U.S.A.;
2. Department of Biological and Environmental Science and Nanoscience Center, University of Jyväskylä, Jyväskylä, Finland;
3. Quest Diagnostics Nichols Institute, San Juan Capistrano, CA, U.S.A.
Received 2010-1-26; Accepted 2010-5-24; Published 2010-5-25
Abstract
A clinical association between idiopathic liver disease and parvovirus B19 infection has been observed. Fulminant liver failure, not associated with other liver-tropic viruses, has been attributed to B19 in numerous reports, suggesting a possible role for B19 components in the extensive hepatocyte cytotoxicity observed in this condition. A recent report by Abe and colleagues (Int J Med Sci. 2007;4:105-9) demonstrated a link between persistent parvovirus B19 genotype I and III infection and fulminant liver failure. The genetic analysis of isolates obtained from these patients demonstrated a conservation of key amino acids in the nonstructural protein 1 (NS1) of the disease-associated genotypes. In this report we examine a conserved residue identified by Abe and colleagues and show that substitution of isoleucine 181 for methionine, as occurs in B19 genotype II, results in the reduction of B19 NS1-induced cytotoxicity of liver cells. Our results support the hypothesis that in the setting of persistent B19 infection, direct B19 NS1-induced cytotoxicity may play a role in idiopathic fulminant liver failure.
Keywords: Parvovirus, B19, Fulminant Liver Failure, Cytotoxicity, Apoptosis
INTRODUCTION
Human parvovirus B19 is a ubiquitous infectious agent known to cause erythema infectiosum in children, bone marrow suppression and chronic anemia in susceptible individuals, and fetal hydrops and death in intrauterine infections (2, 25). Studies have also linked the virus to numerous idiopathic conditions such as systemic lupus erythematosus (SLE)-like syndromes, rheumatoid-like arthritis, and liver and kidney disease (10, 27, 37, 40). The role of B19 in the latter set of pathologies is difficult to discern because the cellular tropism of the virus for erythroid precursor cells (33) excludes viral replication as being the cause of the observed cellular damage in replication nonpermissive tissues. Two major hypotheses have been proposed to explain the potential role of B19 in such phenomena: an immune mediated attack of end-organ antigens due to B19-induced molecular mimicry (22, 43), or direct B19-mediated cellular damage in the absence of viral particle replication (43). Evidence for both mechanisms exists and it is possible that both have a role to play in the diverse clinical picture of B19 disease.
The B19 linear, single-stranded DNA genome encodes three major viral proteins; two structural capsid proteins (VP1 and VP2) and three non-structural proteins (the major 77 kDa NS1 and the minor 11 kDa and 7.5 kDa proteins, the latter two whose functions remain unclear) (39, 48). The humoral response to B19 infection is initially directed against the major viral capsid protein VP2. As the recovery process progresses, however, this response becomes replaced by antibodies against the unique region of VP1 (24). It has been suggested that immune-mediated tissue injury in B19 infection may be the result of cross-reactivity between IgG antibodies against VP epitopes and human antigens (22, 23). On the other hand, the non-structural protein of B19 (NS1) is a candidate mediator of direct cell injury.
B19 virus targets erythroid cells via the major host cell receptor glycolipid globoside, also known as the blood group P antigen (4), as well as two coreceptors, α5β1 and Ku80. The presence of P antigen, however, is not sufficient for B19 replication (43). Studies have suggested that intracellular conditions, such as viral DNA transcription and RNA processing, are involved in establishing cell permissiveness (5, 14, 21). However, tissues identified as non-permissive for B19 replication have been shown to harbor genomic evidence of B19 infection for extended periods of time (7, 40). Although such non-permissive cells display an inability to produce viral structural proteins and replicate the viral genome, curiously these same tissues allow transcription and translation of NS1 (18, 34, 47).
The non-structural NS proteins of parvoviruses are central to the viral life cycle, mediating viral genome transcription, replication, and packaging. These multi-domain proteins have a conserved architecture: the N-terminus encodes for the DNA binding and endonuclease domains (30, 45); the central region contains the conserved NTP-binding and helicase motifs that identify the protein as a member of the AAA+ SF3 helicase group (9, 13, 25); and the unique C-terminus region has been suggested to be involved in host-protein interactions and promoter trans-activation activity (10, 19, 31). The three major genotypes of B19 have been shown to have most of their nucleotide diversity localized to the C-terminus region of its non-structural protein (16, 37). However, until recently no specific amino acid variation has been associated with a single clinical presentation of B19 disease (16, 20). In 2007, Abe and colleagues (1) performed a sequence analysis of B19 NS1 isolated from the livers of two patient groups; group A contained patients with idiopathic fulminant hepatitis while group B was made up of patients with biliary atresia. The study correlated the presence of genotypes 1 and 3 in patients with idiopathic fulminant hepatitis, while only genotype 2 was identified in patients with biliary atresia. Furthermore, conserved (I181M, E201D) amino acid substitutions were identified in patients with biliary atresia that were not observed in the fulminant hepatitis group. Since biliary atresia is a congential condition yet to be linked with an infectious cause (36), identification of conserved amino acid substitutions in the B19 NS1 isolates of only the group A patients suggests that these residues may be critical to the mechanism of B19-associated fulminant liver failure.
B19 has long been linked with a spectrum of liver disease ranging from self-limited hepatitis to acute fulminant liver failure (18, 28, 46). The significant association of bone marrow suppression with acute fulminant liver failure in the pediatric population (6, 15) along with the isolation of B19 DNA from liver tissue (12), which has been shown to be non-permissive for viral replication (3), but allows transcription and translation of B19 NS1 (18, 34), has lead us to examine the potential role of B19 NS1 in direct hepatocyte cytotoxicity. Our laboratory previously demonstrated that B19 NS1 caused apoptosis in liver-derived cells (34, 35). The above described study by Abe et al. (1) led us to explore the effect of the NS1 I181M amino acid substitution on B19 NS1-induced cytotoxicity in liver cells. Our work has shown that the I181M substitution significantly reduces B19 NS1-induced apoptosis in HepG2 cells, suggesting an important role for this residue in the mechanism of B19 NS1-induced liver cytotoxicity.
MATERIALS AND METHODS
Cell Culture. Spodoptera frugiperda-derived SF9 cells were cultured in spinner flasks using BioWhittaker® Insect-XPress cell media (BioWhittaker® , Walkersville, MD) supplemented with 1% Penicillin-Streptomycin (PenStrep) (Gibco®, Invitrogen, Carlsbad, CA) at 27°C. HepG2 cells were cultured in Hepatocyte Wash Media (Gibco®) supplemented with 10% Fetal Bovine Serum (FBS) (Gibco®), 1% L-glutamine (Gibco®) and 1% PenStrep. The cells were incubated at 37°C in 5% CO2.
Cloning and Recombinant Baculovirus Production. A modified pFastBac1 (Invitrogen, Carlsbad, CA) vector was used as a backbone for the generation of B19 NS1 expressing baculovirus. The polyhedrin promoter was removed from the pFastBac1 plasmid by SnaB1 (Fermentas, Glen Burnie, MD) and BamH1 (Fermentas) digestion. The Cytomegalovirus (CMV) immediate-early promoter was obtained from pcDNA3.1 (Invitrogen) by NruI (Fermentas) and BamH1 digestion and used to replace the SnaB1-BamH1 fragment in pFastBac1, resulting in pCMVFastBac1. The vector was again digested with Nhe1 (Fermentas) and BamH1 in order to insert a fragment encoding the Enhanced GFP (EGFP) protein obtained from pEGFP-C1 (Clontech Laboratories Inc., Saint-Germain-en-Laye, France) using the same restriction enzymes, resulting in pCMVEGFPwoFastBac1. The native B19 NS1 sequence was obtained from the PBDP2 vector (34). Mutagenesis of the native sequence was performed using the sequential PCR technique (8). Briefly, two overlapping fragments of the desired mutant construct were produced using unique internal oligonucleotides encoding a collection of point mutations and deletions and B19 NS1 generic external primers (Table 1). The two fragments were then annealed by mutually primed synthesis using the EcoR1 (Fermentas) and Xba1 (Fermentas) encoding B19 NS1 generic external primers (Table 1). The following mutants were generated: PathT, a single amino acid replacement, T183A, and PathI, a single amino acid replacement, I181M. The NS1 constructs were ligated into the EcoR1-Xba1 digested pCMVEGFPwoFastBac1 vector backbone to produce the desired EGFP-NS1 fusion protein and its mutations under the CMV immediate-early promoter.
Primers Used for B19 NS1 and Substitution Construct Cloning.
| Construct | Primer |
|---|---|
| External Primers | F- 5' GGCGACGAATTCATGGAGCTATTTAG GG R- 5' GGCCATCTAGATTACTCATAATCTACAAAGCT |
| PathT - PRMTTA | 1- 5' AGTATCATTTATGGCTACGGTAATG 2- 5' ATTACCGTAGCCATAAATGATACTAGTAG |
Recombinant baculoviruses were prepared using the Bac-to-Bac® Baculovirus Expression system (Invitrogen). Third generation virus was collected by centrifugation at 1500 x g for 10 min. Sterile inactivated fetal bovine serum (FBS) (Gibco®) was added to each virus to make a 10% solution after which the stock was filtered through a 0.2 µm filter (Millipore, Billerica, MA) and stored at 4°C.
Baculovirus Transduction of HepG2 Cells. HepG2 cells were seeded in culture flasks and grown overnight in supplemented media as described above. The following day the media was removed, cells washed once with sterile phosphate buffered saline (PBS), and the baculovirus solution added to the culture flask. A multiplicity of infection (moi) equivalent to ~100-200 was used for each experiment, resulting in a transduction efficiency of ~25% for each virus at 24 hrs post-transduction. The treated HepG2 cells were kept on ice and remained immersed in the baculovirus solution for 1 hr to promote viral binding to the cellular membrane, after which the virus suspension was removed, the cells washed once with sterile PBS and then pre-warmed supplemented media added. Transduced HepG2 cells were stored at 37°C in 5% CO2. Transduction efficiency was established using flow cytometry as described below.
Flow Cytometry. HepG2 cells were collected by trypsinization (0.5% Trypsin EDTA (Gibco®) in PBS (Gibco®) at indicated times, washed once with fresh media (500 µL) and counted to prepare required cell numbers per sample for each experiment. For transduction efficiency analysis, 5 x 105 HepG2 cells were resuspended in 500 µL of ice-cold PBS and immediately analyzed by flow cytometry. For AnnexinV binding analysis 5 x 105 cells were washed once with PBS and resuspended in 500 µL AnnexinV Binding Buffer and 5 µL AnnexinV-PE (AMS Biotechnology LTD., Abington, OX, UK), incubated for 15 min at room temperature in the dark and immediately analyzed by flow cytometry. The percentage of cells expressing the EGFP marker, as well as the distribution of signal intensity for each construct transduced, were established, in order to ensure that a comparable amount of protein was being expressed in each construct transduction, for the comparison of different NS1 mutant groups.
For examination of apoptosis using the SubG1 population in cell cycle analysis, 3 x 106 cells were washed once with ice cold PBS and fixed in 70% ice-cold ethanol (900 μl) for 24-48 hrs at 4ºC. The fixed cells were washed twice with PBS to rehydrate the sample and incubated in propidium iodide 40 μg/ml (Molecular Probes (Invitrogen), Carlsbad, CA) and RNase 40 μg/ml (Roche Basel, Switzerland) in PBS at 37ºC for 20 min. The cells were centrifuged, resuspended in ice-cold PBS and immediately analyzed by flow cytometry.
All samples were analyzed on the FACSCALIBUR flow cytometer (Becton-Dickson, Frankline Lakes, NJ), data collected using Cell-Quest software (Becton-Dickson), and statistical analysis completed by FlowJo software (Tree Star, Inc, Ashland, OR).
Cytotoxicity Assay. Trypan blue dye exclusion was used to determine cell viability. Adherent and loose HepG2 cells were collected by trypsinization (0.5% Trypsin EDTA (Gibco®) in PBS (Gibco®) at indicated times and washed once in sterile PBS. The cells were then resuspended 1:10 in trypan blue solution (0.8 mM trypan blue in PBS) and incubated at room temperature 2-5 min. The cells were counted using a hemocytometer and trypan blue stained cells were identified. The results were quantified as number of trypan blue staining cells out of total cells counted.
Statistical Analysis. Student t-test was used to assess the statistical significance in the data.
RESULTS
Substitution I181M in B19 NS1 interferes with NS1-induced apoptotic cell death. To test the hypothesis that B19-associated liver pathology is related to the direct toxicity of persistent B19 NS1 in hepatocytes of previously infected individuals, we examined the possible role of specific amino acid variation in the B19 NS1 protein on liver toxicity. The clinical observation by Abe et al. (1) of B19 genotype variability in different liver disease groups, identified two amino acid substitutions in B19 NS1 that differed between the biliary atresia and fulminant liver failure patient groups. Substitution E201D, believed to be in the B cell epitope of B19 NS1 (40), substituted one acidic residue for another, suggesting a conserved mutation that would not significantly change the chemical properties of the protein. An immunological analysis of B-cell activation would need to be completed in order to understand the potential impact of this substitution on B19 NS1. Therefore, we focused our analysis on the second substitution, I181M, observed only in genotype 2-carrying biliary atresia patients. We created EGFP tagged constructs of wild type genotype 1, I181M (PathI) substitution and T183A (PathT) substitution of B19 NS1 under the CMV immediate early promoter. The PathT substitution was observed in genotype 3 of B19 but not in genotype 1, both of which could be found in patients with fulminant liver failure, and was used as a second negative control, along with the wild-type B19 NS1 construct. These constructs were cloned into the baculovirus expression system, which was used as a gene delivery vehicle.
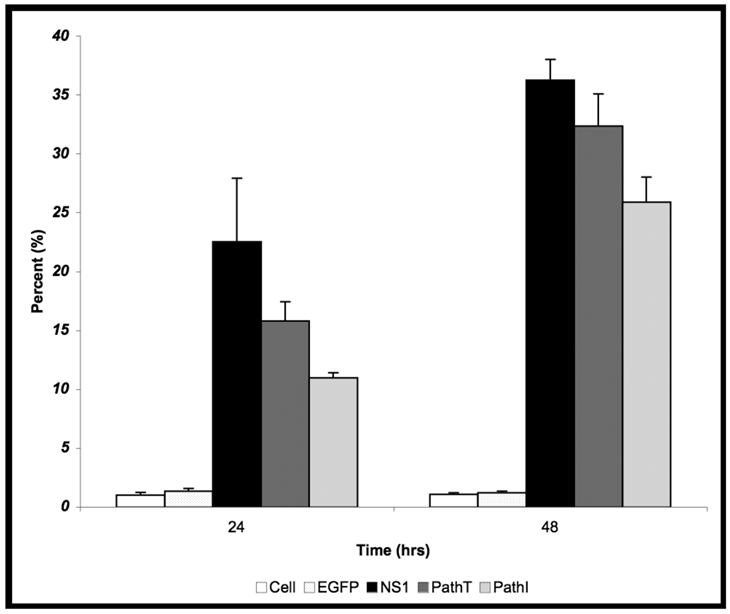
24 hrs post-transduction of HepG2 cells with baculovirus carrying the above described B19 NS1 constructs, trypan blue dye uptake analysis showed an increased amount of cell death in cells expressing B19 NS1-derived proteins compared to mock infected (Cell) or EGFP expressing cells (Figure 1). Although slightly less death could be seen in the PathI transduced cells at 24 hr post-transduction, the difference between the constructs was not statistically significant. HepG2 cell cytotoxicity continued to increase as the cells continued to express B19 NS1. At 48 hrs post-transduction 36% (Figure 1) of HepG2 cells expressing wild-type NS1 showed loss of viability. HepG2 cells transduced with constructs expressing PathI and PathT exhibited a similar pattern of cell loss, with continued decline in cell viability until 48 hrs post-transduction. However, unlike wild-type B19 NS1- and PathT-transduced cells, approximately 10% fewer PathI mutant transduced HepG2 cells showed trypan blue uptake at 48 hrs post-transduction (p = 0.0002) (Figure 1).
Cytotoxicity Assay. HepG2 cells were mock, EGFP-, NS1-, PathT-, or PathI- transduced, collected at indicated times post-transduction and analyzed by trypan blue exclusion. The data represent the mean of three independent experiments demonstrating the percentage of total cells in the sample permeable to trypan blue uptake. One star (*) - statistically significant (p < 0.05) change between Cell and marked set. Two stars (**) - statistically significant (p< 0.05) change between NS1 and marked set. Bars demonstrate the standard deviation between individual runs.
Previous studies have identified apoptosis as the underlying mechanism for the cytotoxicity observed in the setting of transient B19 NS1 expression (34). We therefore examined whether the reduced cell loss observed in the PathI substitution- carrying HepG2 cells resulted from a reduction in cells undergoing apoptosis. We used the AnnexinV binding assay to assess apoptotic cell death in HepG2 cells. The pattern of apoptosis observed in HepG2 cells expressing all B19 NS1 constructs showed continually increasing percentage of cells undergoing apoptosis starting at 24 hrs (Figure 2). At 48 hrs post-transduction the PathI construct expressing cells displayed approximately 18% less apoptotic cell death than the wild-type and PathT B19 NS1-expressing cells (p = 0.023) (Figure 2). The reduced apoptotic cell death in the PathI-expressing cells resembled the reduction observed with the generalized cytotoxicity assay, suggesting that the reduced cell loss is due to a decrease in B19 NS1-induced cell suicide, previously reported to be the mechanism of B19 NS1-induced cytotoxicity (34, 35).
AnnexinV Binding Assay. HepG2 cells were mock, EGFP-, NS1-, PathT-, or PathI-transduced, collected at indicated times post-transduction, and analyzed by flow cytometry for AnnexinV binding. Graphic representation of the means of three independent experiments, demonstrating the percentage of EGFP or EGFP-fusion protein expressing HepG2 cells that bind AnnexinV-PE, as identified by flow cytometry analysis. The AnnexinV results have been normalized for background AnnexinV-PE binding using mock-transduced cells. Bars demonstrate the standard deviation between individual runs. One star (*) - statistically significant (p < 0.05) change between Cell and marked set. Two stars (**) - statistically significant (p< 0.05) change between NS1 and marked set.

Further confirmation for the reduction of apoptosis in the PathI transduced population of HepG2 cells was obtained using cell cycle analysis of HepG2 cells expressing the wild-type and mutant B19 constructs. Non-transduced HepG2 cells, HepG2 cells transduced with moi 400 of non-engineered baculovirus, and HepG2 cells transduced with EGFP expressing virus to 95% transduction efficiency at 24 hrs post-transduction were used to establish HepG2 cell cycle distribution (negative controls). G0/G1, S, G2/M and SubG1 (representing cells undergoing apoptotic cell death) phases were identified on the linear fluorescence intensity plots for propidium iodide emission (Figure 3A). All three negative control conditions displayed the same pattern of cell cycle progression with approximate distribution of cells at 48 hrs as follows; 71% G1, 16% S, 12% G2 and 1% SubG1 (data not shown). For clarity only one representative negative control condition, labeled Cell, is presented in Figure 3.
Cell Cycle Analysis for SubG1 Content. (A) Representative fluorescence intensity plots for propidium iodide emission demonstrating the cell cycle distribution of mock-transduced control (top) and NS1 (bottom) expressing HepG2 cells at 24 and 48 hrs post-transduction. (B) Quantitative graphic representation of HepG2 cell cycle distribution for mock, NS1, PathT and PathI in the SubG1 phase of the cell cycle. The data represents at least two independent experiments with bars demonstrating the standard deviation between the individual runs. One star (*) indicates statistically significant (p < 0.05) change between Cell and marked set. Two stars (**) - statistically significant (p< 0.05) change between NS1 and marked set. Three stars (***) - statistically significant (p< 0.05) change between PathT and marked set.
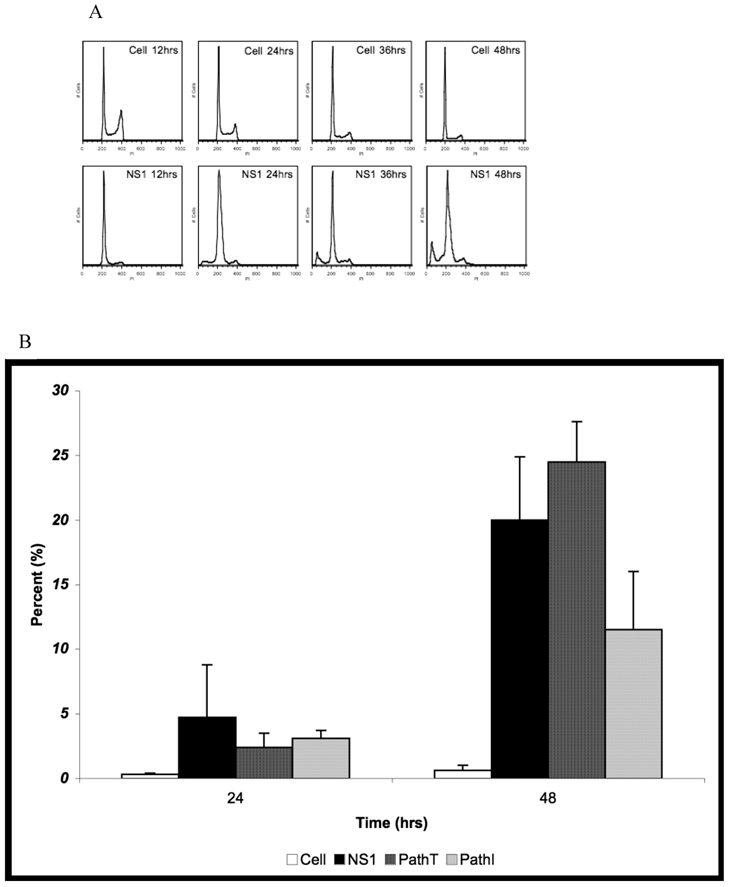
HepG2 cells transduced with baculovirus carrying the NS1 constructs were identified by flow cytometry and subsequent cell cycle analysis performed. The pattern of cell cycle distribution for NS1 expressing cells differed significantly from that seen for the negative controls (Figure 3A bottom panels). At 48 hrs post-transduction 20% of HepG2 cells expressing the NS1 construct were in the SubG1 population (Figure 3B). The SubG1 accumulation demonstrated the cytotoxic nature of B19 NS1, which has been previously shown to cause apoptotic cell death in HepG2 cells.
The cell cycle observations for NS1 expressing cells were then compared to cells expressing the PathT and PathI mutation constructs. At 24 hrs post-transduction all mutant NS1 proteins exhibited a similar effect on HepG2 cells as the wild-type NS1 protein with approximately 3% of cells in the SubG1 population (Figure 3B). At 48 hrs post-transduction, however, a mean of 24.5 % of HepG2 cells expressing the PathT construct were found in the SubG1 population (Figure 3B). In contrast to this, only 11.5% of PathI expressing HepG2 cells were identified in the SubG1 population (Figure 3B), suggesting a significant reduction in apoptotic cell death (p = 0.0317) compared to the PathT construct expressing HepG2 cells. No statistical significance was identified between the wild-type NS1 and PathT construct expressing cells, which we believe is due to the large variability in the SubG1 content data for the wild-type NS1 transduction.
DISCUSSION
Suggestions of a clinical link between B19 and a variety of liver disorders have been reported in the literature (18, 29, 46). However, the mechanism for B19- induced hepatotoxicity remains obscure. Two hypotheses have been proposed: immune-mediated hepatocyte damage resulting from B19-induced molecular mimicry (23, 46), or direct liver toxicity from B19 infection of hepatocytes (34, 46). We have previously shown that B19 can infect liver cells and transcribe and translate its non-structural protein (35). Expression of NS1 in primary hepatocytes as well as in HepG2 cells was shown to be cytotoxic (34). Activation of the innate apoptotic cascade was demonstrated to be the route of cell demise in the setting of B19 NS1 over-expression, suggesting direct NS1-induced cellular damage (35). The endogenous apoptotic cascade is often activated in cells that cannot overcome a set of well defined cellular insults such as an accumulation of DNA damage, disruptions in mitochondrial membrane integrity, or interference with the dynamic cytoskeletal network. Studies with B19 and other parvoviruses have suggested that the parvoviral non-structural protein can potentially interfere with cellular cytoskeleton proteins (31), induce DNA damage (32), and promote mitochondrial apoptotic activation (17). This evidence along with our own observations led us to investigate the role of direct B19 injury to hepatocytes in the setting of B19- associated liver disease.
Parvoviral non-structural proteins are essential for viral replication and packaging, serving numerous roles in the viral life cycle. The multi-domain architecture of parvoviral non-structural proteins results in high conservation of their sequence identity, especially within the well-characterized catalytic regions, such as the endonuclease and NTP-binding motifs. The highly integrated functions of complex proteins can often be interrupted by single, well placed, amino acid disruptions, which can potentially change the native folding, catalytic capacity, oligomerization state, or other inter-protein interactions. The identification by Abe and colleagues (1) of an amino acid substitution in the NS1 protein of B19 genotype 2 that could not be observed in NS1 proteins isolated from livers of patients with fulminant liver failure, prompted us to explore whether this amino acid substitution can effect B19 NS1's cytotoxicity in liver derived cells. In this study, we have demonstrated that substitution I181M significantly reduces genotype 1 B19 NS1's cytotoxicity in HepG2 cells. Furthermore, apoptosis was reduced by the PathI construct of B19 NS1, suggesting that the disruption is in the mechanism by which B19 NS1 induces apoptosis in liver cells.
Due to a lack of structural information about B19 NS1, the role of residue 181 in NS1 function is unclear. The residue does not appear to fall into the clearly identifiable conserved motifs, such as the endonuclease metal coordination motif HuHuu (residues 81-85), or NTP binding p-loop, GXXXXGK (residues 328-334), seen in all parvoviral non-structural proteins. However, even though the primary structure is not telling, the contribution of this residue to the overall structure of the enzymatic folds can be proposed. The change from a hydrophobic residue to a more polar one also suggests a disruption in the potential orientation of the side chain and hence a change in the secondary structure of that region in the protein. Since the I181M substitution did not obliterate B19 NS1 cytotoxicity, but only reduced it, the protein continues to maintain some function, suggesting that this substitution may also reduce viral fitness. B19 replication is difficult to study due to the lack of an efficient B19 viral replicon and therefore direct experimental verification of the role of amino acid variation on replication efficiency or viral fitness must await the availability of such tools. However, based on the results of this study, we can conclude that substitution I181M reduces B19 NS1 cytotoxicity in liver cells, potentially resulting in reduced hepatocyte dropout and thereby a reduced burden of B19-related liver disease.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Abe K, Kiuchi T, Tanaka K, Edamoto Y, Aiba N, Sata T. Characterization of erythrovirus B19 genomes isolated in liver tissues from patients with fulminant hepatitis and biliary atresia who underwent liver transplantation. Int J Med Sci. 2007;4:105-9
2. Anderson M.J, Higgins PG, Davis LR, Willman JS, Jones SE, Kidd IM, Pattison JR, Tyrrell DA. Experimental parvoviral infection in humans. J Infect Dis. 1985;152:257-65
3. Bonvicini F, Filippone C, Manaresi E, Zerbini M, Musiani M, Gallinella G. HepG2 hepatocellular carcinoma cells are a non-permissive system for B19 virus infection. J Gen Virol. 2008;89:3034-8
4. Brown K.E, Anderson SM, Young NS. Erythrocyte P antigen: cellular receptor for B19 parvovirus. Science. 1993;262:114-7
5. Brunstein J, Soderlund-Venermo M, Hedman K. Identification of a novel RNA splicing pattern as a basis of restricted cell tropism of erythrovirus B19. Virology. 2000;274:284-91
6. Cattral M.S, Langnas AN, Markin RS, Antonson DL, Heffron TG, Fox IJ, Sorrell MF, Shaw BWJr. Aplastic anemia after liver transplantation for fulminant liver failure. Hepatology. 1994;20:813-8
7. Corcioli F, Zakrzewska K, Rinieri A, Fanci R, Innocenti M, Civinini R, De Giorgi V, Di Lollo S, Azzi A. Tissue persistence of parvovirus B19 genotypes in asymptomatic persons. J Med Virol. 2008;80:2005-11
8. Cormack B. Current Protocols in Molecular Biology, Supplement 37 ed, vol 1. John Wiley & Sons Inc. 1997
9. Davis M.D, Wu J, Owens RA. Mutational analysis of adeno-associated virus type 2 Rep68 protein endonuclease activity on partially single-stranded substrates. J Virol. 2000;74:2936-42
10. Di Pasquale G, Stacey SN. Adeno-associated virus Rep78 protein interacts with protein kinase A and its homolog PRKX and inhibits CREB-dependent transcriptional activation. J Virol. 1998;72:7916-25
11. Diaz F, Collazos J. Hepatic dysfunction due to parvovirus B19 infection. J Infect Chemother. 2000;6:63-4
12. Eis-Hubinger A.M, Reber U, Abdul-Nour T, Glatzel U, Lauschke H, Putz U. Evidence for persistence of parvovirus B19 DNA in livers of adults. J Med Virol. 2001;65:395-401
13. Gorbalenya A.E, Koonin EV. Helicases: amino acid sequence comparison and structure-function relationships. Curr Opin Struct Biol. 1993;3:419-429
14. Guan W, Cheng F, Yoto Y, Kleiboeker S, Wong S, Zhi N, Pintel DJ, Qiu J. Block to the production of full-length B19 virus transcripts by internal polyadenylation is overcome by replication of the viral genome. J Virol. 2008;82:9951-63
15. Hagler L, Pastore RA, Bergin JJ, Wrensch MR. Aplastic anemia following viral hepatitis: report of two fatal cases and literature review. Medicine (Baltimore). 1975;54:139-64
16. Hemauer A, von Poblotzki A, Gigler A, Cassinotti P, Siegl G, Wolf H, Modrow S. Sequence variability among different parvovirus B19 isolates. J Gen Virol. 1996;77(Pt 8):1781-5
17. Hsu T.C, Wu WJ, Chen MC, Tsay GJ. Human parvovirus B19 non-structural protein (NS1) induces apoptosis through mitochondria cell death pathway in COS-7 cells. Scand J Infect Dis. 2004;36:570-7
18. Karetnyi Y.V, Beck PR, Markin RS, Langnas AN, Naides SJ. Human parvovirus B19 infection in acute fulminant liver failure. Arch Virol. 1999;144:1713-24
19. Legendre D, Rommelaere J. Terminal regions of the NS-1 protein of the parvovirus minute virus of mice are involved in cytotoxicity and promoter trans inhibition. J Virol. 1992;66:5705-13
20. Liefeldt L, Plentz A, Klempa B, Kershaw O, Endres AS, Raab U, Neumayer HH, Meisel H, Modrow S. Recurrent high level parvovirus B19/genotype 2 viremia in a renal transplant recipient analyzed by real-time PCR for simultaneous detection of genotypes 1 to 3. J Med Virol. 2005;75:161-9
21. Liu J.M, Green SW, Shimada T, Young NS. A block in full-length transcript maturation in cells nonpermissive for B19 parvovirus. J Virol. 1992;66:4686-92
22. Loizou S, Cazabon JK, Walport MJ, Tait D, So AK. Similarities of specificity and cofactor dependence in serum antiphospholipid antibodies from patients with human parvovirus B19 infection and from those with systemic lupus erythematosus. Arthritis Rheum. 1997;40:103-8
23. Lunardi C, Tiso M, Borgato L, Nanni L, Millo R, De Sandre G, Severi AB, Puccetti A. Chronic parvovirus B19 infection induces the production of anti-virus antibodies with autoantigen binding properties. Eur J Immunol. 1998;28:936-48
24. Modrow S, Dorsch S. Antibody responses in parvovirus B19 infected patients. Pathol Biol (Paris). 2002;50:326-31
25. Momoeda M, Wong S, Kawase M, Young NS, Kajigaya S. A putative nucleoside triphosphate-binding domain in the nonstructural protein of B19 parvovirus is required for cytotoxicity. J Virol. 1994;68:8443-6
26. Munakata Y, Saito-Ito T, Kumura-Ishii K, Huang J, Kodera T, Ishii T, Hirabayashi Y, Koyanagi Y, Sasaki T. Ku80 autoantigen as a cellular coreceptor for human parvovirus B19 infection. Blood. 2005;106:3449-56
27. Naides S.J. Parvovirus B19 infection. Rheum Dis Clin North Am. 1993;19:457-75
28. Naides S.J, Scharosch LL, Foto F, Howard EJ. Rheumatologic manifestations of human parvovirus B19 infection in adults. Initial two-year clinical experience. Arthritis Rheum. 1990;33:1297-309
29. Nobili V, Vento S, Comparcola D, Sartorelli MR, Luciani M, Marcellini M. Autoimmune hemolytic anemia and autoimmune hepatitis associated with parvovirus B19 infection. Pediatr Infect Dis J. 2004;23:184-5
30. Nuesch J.P, Cotmore SF, Tattersall P. Sequence motifs in the replicator protein of parvovirus MVM essential for nicking and covalent attachment to the viral origin: identification of the linking tyrosine. Virology. 1995;209:122-35
31. Nuesch J.P, Rommelaere J. NS1 interaction with CKII alpha: novel protein complex mediating parvovirus-induced cytotoxicity. J Virol. 2006;80:4729-39
32. Op De Beeck A, Caillet-Fauquet P. The NS1 protein of the autonomous parvovirus minute virus of mice blocks cellular DNA replication: a consequence of lesions to the chromatin? J Virol. 1997;71:5323-9
33. Ozawa K, Kurtzman G, Young N. Replication of the B19 parvovirus in human bone marrow cell cultures. Science. 1986;233:883-6
34. Poole B.D, Karetnyi YV, Naides SJ. Parvovirus B19-induced apoptosis of hepatocytes. J Virol. 2004;78:7775-83
35. Poole B.D, Zhou J, Grote A, Schiffenbauer A, Naides SJ. Apoptosis of liver-derived cells induced by parvovirus B19 nonstructural protein. J Virol. 2006;80:4114-21
36. Rauschenfels S, Krassmann M, Al-Masri AN, Verhagen W, Leonhardt J, Kuebler JF, Petersen C. Incidence of hepatotropic viruses in biliary atresia. Eur J Pediatr. 2009;168:469-76
37. Servant A, Laperche S, Lallemand F, Marinho V, De Saint Maur G, Meritet JF, Garbarg-Chenon A. Genetic diversity within human erythroviruses: identification of three genotypes. J Virol. 2002;76:9124-34
38. Seve P, Ferry T, Koenig M, Cathebras P, Rousset H, Broussolle C. Lupus-like presentation of parvovirus B19 infection. Semin Arthritis Rheum. 2005;34:642-8
39. Shade R.O, Blundell MC, Cotmore SF, Tattersall P, Astell CR. Nucleotide sequence and genome organization of human parvovirus B19 isolated from the serum of a child during aplastic crisis. J Virol. 1986;58:921-36
40. Soderlund-Venermo M, Hokynar K, Nieminen J, Rautakorpi H, Hedman K. Persistence of human parvovirus B19 in human tissues. Pathol Biol (Paris). 2002;50:307-16
41. Tolfvenstam T, Lundqvist A, Levi M, Wahren B, Broliden K. Mapping of B-cell epitopes on human parvovirus B19 non-structural and structural proteins. Vaccine. 2000;19:758-63
42. Waldman M, Kopp JB. Parvovirus B19 and the kidney. Clin J Am Soc Nephrol. 2007;2(Suppl 1):S47-56
43. Weigel-Kelley K.A, Yoder MC, Srivastava A. Recombinant human parvovirus B19 vectors: erythrocyte P antigen is necessary but not sufficient for successful transduction of human hematopoietic cells. J Virol. 2001;75:4110-6
44. Weigel-Kelley K.A, Yoder MC, Srivastava A. Alpha5beta1 integrin as a cellular coreceptor for human B19: requirement of functional activation of beta1 integrin for viral entry. Blood. 2003;102:3927-33
45. Yoon M, Smith DH, Ward P, Medrano FJ, Aggarwal AK, Linden RM. Amino-terminal domain exchange redirects origin-specific interactions of adeno-associated virus rep78 in vitro. J Virol. 2001;75:3230-9
46. Yoto Y, Kudoh T, Haseyama K, Suzuki N, Chiba S. Human parvovirus B19 infection associated with acute hepatitis. Lancet. 1996;347:868-9
47. Zakrzewska K, Cortivo R, Tonello C, Panfilo S, Abatangelo G, Giuggioli D, Ferri C, Corcioli F, Azzi A. Human parvovirus B19 experimental infection in human fibroblasts and endothelial cells cultures. Virus Res. 2005;114:1-5
48. Zhi N, Mills IP, Lu J, Wong S, Filippone C, Brown KE. Molecular and functional analyses of a human parvovirus B19 infectious clone demonstrates essential roles for NS1, VP1, and the 11-kilodalton protein in virus replication and infectivity. J Virol. 2006;80:5941-50
Author contact
![]() Corresponding author: Stanley J. Naides, M.D., Mail: 33608 Ortega Highway, San Juan Capistrano, CA 92675. Phone: 949 728-4578; FAX: 949 728-7852; E-mail: stanley.j.naidescom
Corresponding author: Stanley J. Naides, M.D., Mail: 33608 Ortega Highway, San Juan Capistrano, CA 92675. Phone: 949 728-4578; FAX: 949 728-7852; E-mail: stanley.j.naidescom