Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2009; 6(2):85-92. doi:10.7150/ijms.6.85 This issue Cite
Research Paper
Induction and effector phase of allergic lung inflammation is independent of CCL21/CCL19 and LT-beta
Corinne Ploix1, Riaz I. Zuberi2, Fu-Tong Liu3, Monica J. Carson4, David D. Lo4 ![]()
1. Roche Ltd., Basel, Switzerland
2. La Jolla Institute for Molecular Medicine, La Jolla, CA, USA
3. Department of Dermatology, School of Medicine, University of California, Davis, CA, USA
4. Division of Biomedical Sciences, University of California, Riverside, CA, USA
Received 2009-2-17; Accepted 2009-3-6; Published 2009-3-10
Abstract
The chemokines CCL21 and CCL19, and cell bound TNF family ligand lymphotoxin beta (LTβ), have been associated with numerous chronic inflammatory diseases. A general role in chronic inflammatory diseases cannot be assumed however; in the case of allergic inflammatory disease, CCL21/CCL19 and LTβ have not been associated with the induction, recruitment, or effector function of Th2 cells nor dendritic cells to the lung. We have examined the induction of allergic inflammatory lung disease in mice deficient in CCL21/CCL19 or LTβ and found that both kinds of mice can develop allergic lung inflammation. To control for effects of priming differences in knockout mice, adoptive transfers of Th2 cells were also performed, and they showed that such effector cells had equivalent effects on airway hyper-responsiveness in both knockout background recipients. Moreover, class II positive antigen presenting cells (B cells and CD11c+ dendritic cells) showed normal recruitment to the peribronchial spaces along with CD4 T cells. Thus, the induction of allergic responses and recruitment of both effector Th2 cells and antigen presenting cells to lung peribronchial spaces can develop independently of CCL21/CCL19 and LTβ.
Keywords: asthma, dendritic cell, Th2, chemokine
Introduction
The membrane-bound cytokine lymphotoxin beta (LTβ) is known to be important in the early stages of secondary lymphoid tissue development. Signaling by LTα1/β2 ligands through the LTβ-R induces development of the stromal cells of secondary lymphoid tissues [1]. The stromal cells provide organization to these lymphoid tissues in large part by producing the chemotactic CCR7 ligand CCL21, which recruits both lymphocytes and dendritic cells, signaling through the chemokine receptor CCR7 [2,3]. LTβ-deficient mice show greatly impaired immune responses [4-6]; likewise, immune responses are significantly delayed in CCL21 deficient mice [7]. The tissue site and mechanism of induction of immune responses in these mutant mice remain unclear.
Interestingly, several acute and chronic inflammatory diseases also appear to have a strong dependence on LTβ and CCL21 during their effector phase. Thus, blockade of LTβ-R will significantly inhibit graft-versus-host-disease [8,9], experimental colitis [10,11], and autoimmune diabetes [12]. Similarly, strong local CCL21 induction was found to be associated with autoimmune diabetes [13], chronic liver inflammation [14,15], multiple sclerosis [16] and Experimental Autoimmune Encephalomyelitis [17,18], rheumatoid arthritis [19], Sjogren's syndrome [20], and inflammatory skin diseases [21-25]. In addition, CCL21 appears to have a role in regulating CD4 T cell homeostatic proliferation and effector cell development; an excess of CCL21 was found to be sufficient to drive autoimmune diabetes [26]. Most of the inflammatory responses described above arguably fall into the category of Th1-mediated effector mechanisms. Moreover, they are often associated with local development of lymphoid follicle structures (e.g., Sjogren's syndrome, rheumatoid arthritis, autoimmune diabetes) in a process that resembles LTβ- and CCL21-dependent secondary lymphoid tissue development (“lymphoid neo-organogenesis,” [27]).
Thus, to determine whether a similar dependence on LTβ and CCL21 is present in Th2-mediated effector mechanisms in the lung, we studied the induction of allergic lung inflammation in LTβ and CCL21/CCL19 deficient mice. We found that both kinds of mutant mice mounted normal or enhanced Th2 responses as measured by eosinophil recruitment and increased airway resistance. Recruitment of CD4 T cells, B lymphocytes, and CD11c+ dendritic cells to the peribronchial space was also normal. Our results show that the initiation and effector phase of allergic lung inflammation is independent of LTβ and CCL21.
Materials and Methods
Mice
LTβ knockout mice (“LTβ-KO”) backcrossed to C57BL/6 were kindly provided by Dr. S. A. Nedospasov [1], and plt/plt (“plt”) mutant mice which lack expression of the CCR7 ligand chemokines CCL21 and CCL19 [2], backcrossed to BALB/c, were generously provided by Dr. A. Matsusawa. Mice were bred and maintained under specific pathogen-free conditions in accordance with NIH and institutional guidelines.
For the adoptive transfer studies in Figure 3B, adoptively transferred CD4 T cells were taken from donors matching the genetic background of the recipient. Thus, C57BL/6 mice were used as donor of CD4 T cells for C57BL/6 and LTβ-KO mice while plt recipients were injected with cells from BALB/c mice. Transfer recipients were 6-8 weeks old. Although the mutant mice studied were on two different backgrounds, the strain specific controls (e.g., challenged with PBS) were used in each case. In addition, we found that control mice on C57BL/6 and BALB/c background strains could both be induced to develop robust allergic lung inflammation.
Induction of allergic lung inflammation
Naïve mice were injected i.p. at day 0 and day 7 with 10 µg chicken egg ovalbumin (Sigma) in 100 µl of PBS mixed with 100 µl of Imject Alum (Pierce, Rockford, IL). One week later, immunized mice were challenged intra-nasally with 25 µl OVA (2 mg/ml) or PBS once per day for 3 consecutive days.
For adoptive transfer experiments, naïve mice were injected i.p. at day 0 and day 7 with 10 µg chicken egg ovalbumin (Sigma) in 100 µl of PBS mixed with 100 µl of Imject Alum (Pierce, Rockford, IL). One week later, immunized mice were challenged intra-nasally with 25 µl OVA (2 mg/ml) or PBS once per day for 3 consecutive days. Splenic CD4 T cells were purified on day 14 using magnetic beads and were i.v. injected (5x106 cells/mouse). The phenotype of the donor CD4 Th2 cells was confirmed by real-time PCR analysis of RNA for IL-2, IL-4, IFNγ, and HPRT, and calculating the IL-4/IFNγ ratios. Intranasal challenge was performed on day 14, 15 and 16 (days 0, 1, and 2 after transfer) as described previously [28,29].
T cell transfer
Spleens were aseptically removed and single cell suspensions were prepared by mechanical dispersion according to standard procedures in Hanks' balanced salt solution (HBSS). CD4 T cell enriched populations were obtained by elimination of B cells and macrophages by incubating for 30 min on ice with rat anti mouse CD45R/B220, anti-CD8, M5/114 and F4/80 antibodies (PharMingen, San Diego, CA). Cell suspensions were then incubated for 30 min on ice under constant agitation with magnetic beads coated with a sheep anti-rat IgG covalently linked Abs (Dynabeads M-450, Dynal, Oslo, Norway) at a bead to cell ratio of 20:1. Non-adherent cells were collected after the removal of free and cell bound beads using a magnetic bead concentrator (Dynal, MPC 6). The resulting cell populations comprised more than 90% CD4+ cells by flow cytofluorometry using a FITC-labeled anti-mouse CD4 antibody (Pharmingen) on a FACScan flow cytometer (Becton Dickinson, Mountain View, CA). Five million splenic CD4 T cells from donor mice were injected i.v.
Bronchoalveolar lavage (BAL)
BAL was performed 3 hours after the last i.n. challenge using 3x 1ml of RPMI 2% FCS. Cells were counted and resuspended at a final concentration of 5x105 cells/ml. Cytospin slides were fixed with methanol and stained with Diff-Quick (Fisher Scientific, Pittsburgh, PA). Eosinophils, monocytes/macrophages and lymphocytes were enumerated based on morphology and staining and expressed as percentages of total BAL cells [28,29].
Histology
Frozen lung sections were fixed in 1% formamide-acetone and stained for cyanide-resistant peroxidase activity as previously described [28,29]. Other sections were stained with rat anti-mouse CD4 (PharMingen) followed by biotin F(ab')2 mouse anti-rat IgG (Jackson Immunoresearch, West Grove, PA) and streptavidin-FITC (Jackson). APCs, dendritic cells and B lymphocytes were visualized using respectively M5/114, anti-CD11c and anti-B220 Abs (culture supernatant and PharMingen) followed by biotinylated secondary Ab and streptavidin-rhodamine or streptavidin-FITC. For dendritic cells, the signal was amplified using TSA-indirect kit according to manufacturer's instructions (NEN Research Products, Boston, MA) and revealed by streptavidin-Rhodamine (Jackson Immunoresearch).
Airway reactivity measurements
Airway hyper-responsiveness (AHR) to metacholine (Acetyl-β methylcholine chloride, Sigma) was assessed 24 hours after the last intranasal challenge. Briefly, mice were maintained inside a whole-body plethysmograph (Buxco, Troy, NY) capable of measuring changes in airflow and trans-thoracic pressures and resistances [30]. After establishing a stable baseline for total lung resistance, mice were exposed to aerosols of saline or metacholine in increasing concentrations (1 to 50 mg/ml) for 3 min. Enhanced pause (Penh) index values reflecting changes in the waveform of pressure signal from both inspiration and expiration combined with the timing were calculated and averaged for 3 min after each nebulization.
Results
Allergic lung inflammation in mutant mice
In previous studies, it was found that robust lung eosinophilia could be rapidly induced in mice, requiring only the adoptive transfer of a small number of antigen specific Th2 cells combined with antigen challenge [28, 31-33]. These results suggested that lung allergic inflammation was not dependent on any “conditioning” of the lung by antigen specific effector cells (e.g., expressing LTβ), prior antigen exposure, or the presence of allergen specific IgE. Moreover, the rapid kinetics of the inflammation suggested that presentation of antigen in the lung parenchyma might be sufficient to trigger the cascade of cytokines and chemokines necessary for tissue eosinophilia without involvement of draining lymphoid tissues.
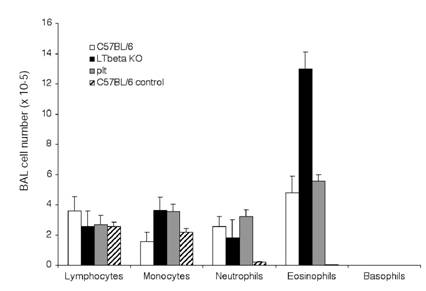
We studied the induction of lung allergic inflammation in mice lacking LTβ (lymphotoxin-beta knockout, or LTβ-KO [1]) or CCL21/CCL19 (plt mutant [2]). In initial studies, control, LTβ-KO, and plt mutant mice were immunized with OVA according to a protocol previously established to induce allergic lung inflammation in normal mice [28,29]. Figure 1 shows that robust lung eosinophilia was generated in all mice at equivalent levels (though possibly increased in LTβ-KO mice), suggesting that both Th2 priming and lung eosinophilia were not impaired by either the LTβ or CCL21/CCL19 defects. The histology of eosinophil infiltration was also unaffected in the mutant mice, as dramatic peribronchial and perivascular infiltration was produced in all mice (Figure 2).
Eosinophilia in BAL: C57BL/6J (white bars), LTβ-KO (black bars) and plt/plt mice (grey bars), were immunized and challenged i.n. with OVA as described in material and methods section. Three hours after the last i.n. injection lung cells were harvested by lavage, counted and submitted to cytospin for analysis of the composition of the BAL. Control C57BL/6J mice (hatched bars) were not immunized but were challenged i.n. with OVA. Results summarize 4 independent experiments with 3 mice per group (12 mice total for each condition).
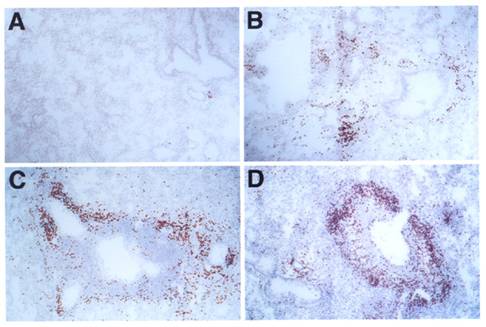
Histology of lung infiltrates: C57BL/6J mice (A, B), LTβ-KO (C) and plt/plt mice (D) were i.p. immunized and i.n. challenged with OVA (B, C, D) or PBS (A). Lungs show peribronchial and perivascular accumulation of eosinophils in mice challenged with OVA but not in those challenged with PBS. Sections were stained with DAB in the presence of cyanide to reveal eosinophil peroxidase activity. Slides were counterstained with H&E. All pictures are at the same magnification (originally photographed at 200x).
Airway resistance in mutant mice
Earlier work suggested that the peribronchial accumulation of eosinophils was due to local T cell production of the cytokines IL-4 and IL-13, which in turn induced epithelial production of the eosinophil chemokine eotaxin [28]. Since local Th2 cell production of cytokines has also been implicated in the induction of increased airway resistance [31,32], we also tested the mice for airway resistance changes.
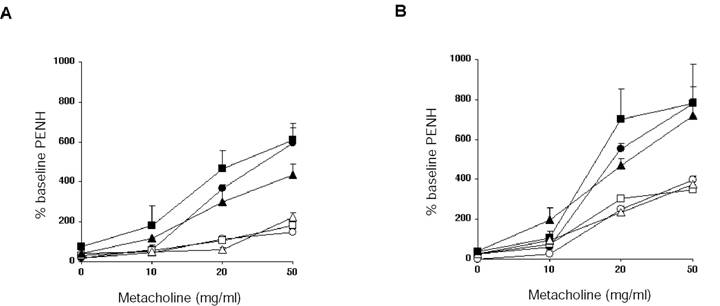
Figure 3 shows the results of airway resistance changes in mice as measured in a whole body plethysmograph. Mice were challenged with increasing amounts of methylcholine, and calculated Penh values were used as an estimate of airway resistance. We found that Penh values were equally increased in controls, LTβ-KO, and plt mutant mice. Moreover, when antigen specific Th2 cells were transferred from wild type mice to naïve mutant mice, increases in airway resistance were also increased to levels exhibited in wild type mice. Thus, Th2 effector responses leading to changes in airway resistance were also not dependent on either LTβ nor CCL21/CCL19.
Airway hyper-reactivity: Twenty-four hours after the last i.n. challenge, airway responsiveness was assessed in i.p. immunized mice (A) and recipients of antigen specific CD4 T cell transfer (B). Airway sensitivity of C57BL/6J mice (circles), LTβ-KO (squares) and plt/plt mice (triangles) was tested in the presence of increasing concentration of metacholine. Mice challenged i.n. with OVA (black symbols) had significantly greater responsiveness to metacholine than their respective PBS controls (white symbols). Results are shown for 3 mice per group, representing one of two independent experiments.
Antigen presenting cell recruitment in the lungs of mutant mice
Antigen presenting cells or their precursors are generally present within nearly all tissues, presumably serving a kind of sentinel function. Inflammatory stimuli such as TNF and endotoxin are extremely effective in inducing resident tissue antigen presenting cells to become activated antigen presenting cells, which then migrate to draining lymph nodes to stimulate cellular immune responses [34]. The migration of activated antigen presenting cells to the draining lymph nodes is considered to be in large part dependent on CCL21-dependent chemotaxis [2,3]. In addition, while LTβ-KO mice have grossly normal dendritic cell subsets [5, 35], immune responses can be impaired, perhaps due in part to defects in homing or recruitment of dendritic cells. Thus it was possible that LTβ or CCL21 deficient mice would show altered recruitment of antigen presenting cells to the lung parenchyma.
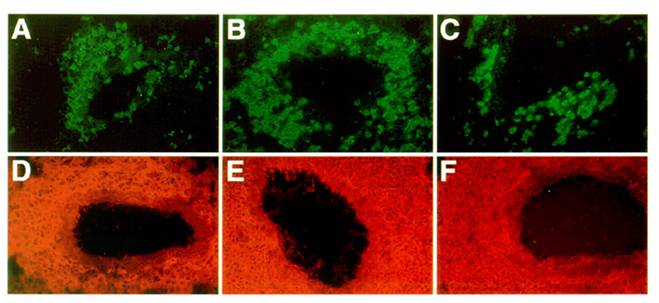
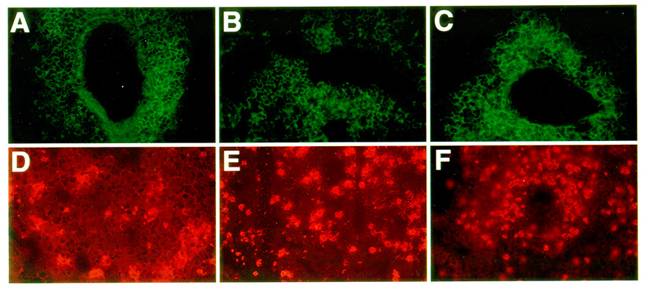
To examine the recruitment of candidate antigen presenting cells, we studied the presence and distribution of class II MHC positive cells within the sites of inflammation in the lung as previously described [36]. Figure 4 shows that class II MHC positive cells are abundant within the perivascular and peribronchial spaces of inflamed lung tissue in both control and mutant mice. In these infiltrates, eosinophils are also present, along with numerous CD4 T cells. However, while infiltrates in some immune mediated inflammatory diseases show spontaneous organization into distinct T cell and B cell dependent compartments, allergic lung infiltrates were rather disorganized.
Organization of infiltrates in the lungs of asthmatic mice: Frozen lung sections from OVA challenged C57BL/6J (A, D), LTβ-KO (B, E) and plt/plt mice (C, F) were stained for antigen presenting cell and T cell markers. In all 3 groups of mice, an accumulation of CD4 T cells (A, B, C) and MHC II positive cells (D, E, F) was observed within the lung parenchyma. All pictures are at the same magnification (originally photographed at 200x).
Next, we examined the lung infiltrates for the character and distribution of two main candidate antigen presenting cell types: namely, B lymphocytes (B220 positive cells) and dendritic cells (CD11c positive cells [37,38]). As shown in Figure 5, control, LTβ-KO, and plt mutant mice all showed similar accumulations of B220 positive B cells and CD11c positive dendritic cells. In sum, it is clear that the recruitment of both antigen presenting cell types remains unimpaired by the absence of LTβ and CCL21.
Nature of the APCs in the lung infiltrates of asthmatic mice: C57BL/6J (A, D), LTβ-KO (B, E) and plt/plt mice (C, F) showed no significant differences in the type and distribution of the antigen presenting cells recruited into the inflammatory tissue. B220 staining revealed a strong perivascular and peribronchial accumulation of B cells (A, B, C) while dendritic cells (CD11c positive cells) tend to be distributed throughout the parenchyma (D, E, F). All pictures are at the same magnification (originally photographed at 200x).
Discussion
The present study was initiated to address two related questions on the mechanisms of immune effector responses. First, a number of chronic inflammatory immune responses show spontaneous organization of infiltrating lymphocytes into follicles with distinct T and B cell compartments, and these are associated with secondary lymphoid tissue factors LTβ and CCL21. Lung allergic inflammation might be considered similar in many respects to chronic inflammation, due to its accumulation of lymphocytic infiltrates and long term induction of tissue remodeling. Therefore, we wanted to determine whether there were elements of the lung allergic inflammatory response that also showed dependence on LTβ and CCL21. Indeed, the formation of germinal centers within the parenchyma has been reported in inflamed lung after airway antigenic challenge [39]. Our results show that the cellular infiltrates in allergic lung inflammation are organized only to the extent that they are limited to the peribronchial and perivascular spaces. There is recruitment of T and B lymphocytes as well as dendritic cells, but there does not appear to be any consistent organization within the infiltrates themselves, suggesting that the pathogenesis of bronchial asthma is not dependent on germinal center formation. Moreover, there is no evidence that organized secondary lymphoid tissues draining the lung parenchyma are absolutely required; one recent study demonstrated that an intact spleen is sufficient for the generation of Th2 responses in the lung even in the absence of secondary lymphoid tissue (in LTα knockout mice [40]). Thus, allergic lung inflammation can be generated independently of organized secondary lymphoid tissue and the associated factors LTβ and CCL21.
Second, studies have suggested that chemokines such as CCL21 are important to the development of cellular immunity, as CCL21 helps in the recruitment of antigen presenting cells, especially dendritic cells, to lymphoid tissue [2,3,34] and in chronic inflammation [13]. Our results suggest that in the case of allergic lung responses, CCL21 is not required for recruitment of dendritic cells. It is likely that other cytokines and chemokines are still important in recruitment of the infiltrating cells [41], assisted by the induction of adhesion molecules such as ICAM-1 and VCAM-1. The accumulation of both T cells and antigen presenting cells into the peribronchial and perivascular spaces may be sufficient to allow for effective local antigen presentation simply by bringing the cells together in a single confined compartment.
Recent studies have also examined the ability of CCL21/CCL19-deficient mice to develop allergic inflammation [42-45], though we are not aware of similar studies on mice lacking LTβ. Initial studies on the CCL21/CCL19-deficient plt mouse strain had shown that immunologic priming showed a delayed but in some cases enhanced response [7], so depending on the background strain and timing of the experimental protocols, one might expect to find reduced or enhanced responses. Indeed, in the various published reports, both reduced and enhanced responses have been reported in plt mice, leading to contradicting conclusions on the importance of CCL21/CCL19 ligands in allergic inflammation. In our studies, we also incorporated adoptive transfer of established Th2 cells from normal donors to show that differentiated effector cells showed equivalent effects on airway hypersensitivity regardless of the presence of CCL21/CCL19, or LTβ.
Based on our results, Th2 responses appear to be independent of the factors associated with lymphoid tissues. Thus, in many cases, local Th2 responses could provide a default response to various immune triggers in the absence of strong Th1 stimuli [46]. In the context of mucosal “tolerance,” responses to antigens presented by antigen presenting cells in the lung (or other mucosal tissues) may be predisposed toward less destructive Th2 responses instead of pathological Th1 responses. The development of pathological Th2 responses seen in allergic lung inflammation may further depend on additional factors, such as sporadic antigen exposure, or other factors unique to the lung anatomy.
Acknowledgements
The authors acknowledge the generosity of Dr. A. Matsuzawa for providing the plt mutant mice and Dr. S. Nedospasov for providing the LTβ knockout mice. This work was supported by grants from the USPHS to M.J.C. and D.L, and RO1 AI-39620 to F.T.L. C.P. was supported by a post-doctoral fellowship from the Fondation pour la Recherche Medicale.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Alimzhanov MB, Kuprash DV, Kosco-Vilbois MH, Luz A, Turetskaya RL, Tarakhovsky A, Rajewsky K, Nedospasov SA, Pfeffer K. Abnormal development of secondary lymphoid tissues in lymphotoxin beta-deficient mice. Proc Natl Acad Sci USA. 1997;94:9302-7
2. Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189:451-60
3. Cyster JG. Chemokines and the homing of dendritic cells to the T cell areas of lymphoid organs. J Exp Med. 1999;189:447-50
4. Lucas R, Tacchini-Cottier F, Guler R, Vesin D, Jemelin S, Olleros ML, Marchal G, Browning JL, Vassalli P, Garcia I. A role for lymphotoxin beta receptor in host defense against Mycobacterium bovis BCG infection. Eur J Immunol. 1999;29:4002-10
5. Berger DP, Naniche D, Crowley MT, Koni PA, Flavell RA, Oldstone MB. Lymphotoxin-beta-deficient mice show defective antiviral immunity. Virology. 1999;260:136-47
6. Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59-70
7. Mori S, Nakano H, Aritomi K, Wang CR, Gunn MD, Kakiuchi T. Mice lacking expression of the chemokines CC21-ser and CCL19 (plt mice) demonstrate delayed but enhanced T cell immune responses. J Exp Med. 2001;193:207-18
8. Tamada K, Tamura H, Flies D, Fu YX, Celis E, Pease LR, Blazar BR, Chen L. Blockade of LIGHT/LTbeta and CD40 signaling induces allospecific T cell anergy, preventing graft-versus-host disease. J Clin Invest. 2002;109:549-57
9. Wu Q, Fu YX, Sontheimer RD. Blockade of lymphotoxin signalin inhibits the clinical expression of murine graft-versus-host skin disease. J Immunol. 2004;172:1630-6
10. Mackay F, Browning JL, Lawton P, Shah SA, Comiskey M, Bhan AK, Mizoguchi E, Terhorst C, Simpson SJ. Both the lymphotoxin and tumor necrosis factor pathways are involved in experimental murine models of colitis. Gastroenterology. 1998;115:1464-75
11. Stopfer P, Obermeier F, Dunger N, Falk W, Farkas S, Janotta M, Moller A, Mannel DN, Hehlgans T. Blocking lymphotoxin-beta receptor activation diminishes inflammation via reduced mucosal addressin cell adhesion molecule-1 (MAdCAM-1) expression and leucocyte margination in chronic DSS-induced colitis. Clin Exp Immunol. 2004;136:21-9
12. Wu Q, Salomon B, Chen M, Wang Y, Hoffman LM, Bluestone JA, Fu YX. Reversal of spontaneous autoimmune insulitis in nonobese diabetic mice by soluble lymphotoxin receptor. J Exp Med. 2001;193:1327-32
13. Hjelmstrom P, Fjell J, Nakagawa T, Sacca R, Cuff CA, Ruddle NH. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am J Pathol. 2000;156:1133-8
14. Grant AJ, Goddard S, Ahmed-Choudhury J, Reynolds G, Jackson DG, Briskin M, Wu L, Hubscher SG, Adams DH. Hepatic expression of secondary lymphoid chemokine (CCL21) promotes the development of portal-associated lymphoid tissue in chronic inflammatory liver disease. Am J Pathol. 2002;160:1445-55
15. Bonacchi A, Petrai I, Defranco RM, Lazzeri E, Annunziato F, Efsen E, Cosmi L, Romagnani P, Milani S, Failli P, Batignani G, Liotta F, Laffi G, Pinzani M, Gentilini P, Marra F. The chemokine CCL21 modulates lymphocyte recruitment and fibrosis in chronic hepatitis C. Gastroenterology. 2003;125:1060-76
16. Pashenkov M, Soderstrom M, Link H. Secondary lymphoid organ chemokines are elevated in the cerebrospinal fluid during central nervous system inflammation. J Neuroimmunol. 2003;135:154-60
17. Alt C, Lashinger M, Engelhardt B. Functional expression of the lymphoid chemokines CCL19 (ELC) and CCL21 (SLC) at the blood-brain barrier suggests their involvement in G-protein-dependent lymphocyte recruitment into the central nervous system during experimental autoimmune encephalomyelitis. Eur J Immunol. 2002;32:2133-44
18. Columba-Cabezas S, Serafini B, Ambrosini E, Aloisi F. Lymphoid chemokines CCL19 and CCL21 are expressed in the central nervous system during experimental autoimmune encephalomyelitis: implications for the maintenance of chronic neuroinflammation. Brain Pathol. 2003;13:38-51
19. Weyand CM, Goronzy JJ. Ectopic germinal center formation in rheumatoid synovitis. Ann NY Acad Sci. 2003;987:140-9
20. Salomonsson S, Jonsson MV, Skarstein K, Brokstad KA, Hjelmstrom P, Wahren-Herlenius M, Jonsson R. Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren's syndrome. Arthritis Rheum. 2003;48:3187-201
21. Christopherson 2nd KW, Hood AF, Travers JB, Ramsey H, Hromas RA. Endothelial induction of the T-cell chemokine CCL21 in T-cell autoimmune diseases. Blood. 2003;101:801-6
22. Mitsui G, Mitsui K, Hirano T, Ohara O, Kato M, Niwano Y. Kinetic profiles of sequential gene expressions for chemokines in mice with contact hypersensitivity. Immunol Lett. 2003;86:191-7
23. Eberhard Y, Ortiz S, Ruiz Lascano A, Kuznitzky R, Serra HM. Up-regulation of the chemokine CCL21 in the skin of subjects exposed to irritants. BMC Immunol. 2004;5:7
24. Lew W, Bowcock AM, Krueger JG. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol. 2004;25:295-305
25. Serra HM, Eberhard Y, Martin AP, Gallino N, Gagliardi J, Baena-Cagnani CE, Lascano AR, Ortiz S, Mariani AL, Uguccioni M. Secondary lymphoid tissue chemokine (CCL21) is upregulated in allergic contact dermatitis. Int Arch Allergy Immunol. 2004;133:64-71
26. Ploix C, Lo D, Carson MJ. A ligand for the chemokine receptor CCR7 can influence the homeostatic proliferation of CD4 T cells and progression of autoimmunity. J Immunol. 2001;167:6724-6730
27. Ruddle NH. Lymphoid neo-organogenesis: lymphotoxin's role in inflammation and development. Immunol Res. 1999;19:119-25
28. Li L, Xia Y, Nguyen A, Feng L, Lo D. Th2-induced eotaxin expression and eosinophilia coexist with Th1 responses at the effector stage of lung inflammation. J Immunol. 1998;161:3128-35
29. Li L, Crowley M, Nguyen A, Lo D. Ability of a nondepleting anti-CD4 antibody to inhibit Th2 responses and allergic lung inflammation is independent of coreceptor function. J Immunol. 1999;163:6557-66
30. Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, Gelfand EW. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156:766-75
31. Hansen G, Berry G, DeKruyff RH, Umetsu DT. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest. 1999;103:175-83
32. Cohn L, Tepper JS, Bottomly K. IL-4-independent induction of airway hyperresponsiveness by Th2, but not Th1, cells. J Immunol. 1998;161:3813-6
33. Randolph DA, Stephens R, Carruthers CJ, Chaplin DD. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J Clin Invest; 104: 1021-9.
34. Sallusto F, Lanzavecchia A. Mobilizing dendritic cells for tolerance, priming, and chronic inflammation. J Exp Med. 1999;189:611-4
35. Crowley M, Lo D. Targeted gene knockouts: Insights into dendritic cell biology. In: (ed.) Lotze M.T, Thomson A.W. Dendritic cells: Biology and clinical application. San Diego: Academic Press. 1999:579-593
36. McWilliam AS, Napoli S, Marsh AM, Pemper FL, Nelson DJ, Pimm CL, Stumbles PA, Wells TN, Holt PG. Dendritic cells are recruited into the airway epithelium during the inflammatory response to a broad spectrum of stimuli. J Exp Med. 1996;184:2429-32
37. Holt PG, Haining S, Nelson DJ, Sedgwick JD. Origin and steady-state turnover of class II MHC-bearing dendritic cells in the epithelium of the conducting airways. J Immunol. 1994;153:256-61
38. Schon-Hegrad MA, Oliver J, McMenamin PG, Holt PG. Studies on the density, distribution, and surface phenotype of intraepithelial class II major histocompatibility complex antigen (Ia)-bearing dendritic cells (DC) in the conducting airways. J Exp Med. 1991;173:1345-56
39. Chvatchko Y, Kosco-Vilbois MH, Herren S, Lefort J, Bonnefoy JY. Germinal center formation and local immunoglobulin E (IgE) production in the lung after an airway antigenic challenge. J Exp Med. 1996;184:2353-60
40. Gajewska BU, Alvarez D, Vidric M, Goncharova S, Stampfli MR, Coyle AJ, Gutierrez-Ramos J-C, Jordana M. Generation of experimental allergic airways inflammation in the absence of draining lymph nodes. J Clin Invest. 2001;108:577-583
41. Mathew A, Medoff BD, Carafone AD, Luster AD. Cutting edge: Th2 cell trafficking into the allergic lung is dependent on chemoattractant receptor signaling. J Immunol. 2002;169:651-655
42. Yamashita N, Tashimo H, Matsuo Y, Ishida H, Yoshiura K, Sato K, Yamashita N, Kakiuchi T, Ohta K. Role of CCL21 and CCL19 in allergic inflammation in the ovalbumin-specific murine asthmatic model. J Allergy Clin Immunol. 2006;117:1040-6
43. Grinnan D, Sung SS, Dougherty JA, Knowles AR, Allen MB, Rose CE 3rd, Nakano H, Gunn MD, Fu SM, Rose CE Jr. Enhanced allergen-induced airway inflammation in paucity of lymph node T cell (plt) mutant mice. J Allergy Clin Immunol. 2006;118:1234-41
44. Xu B, Aoyama K, Kusumoto M, Matsuzawa A, Butcher EC, Michie SA, Matsuyama T, Takeuchi T. Lack of lymphoid chemokines CCL19 and CCL21 enhances allergic airway inflammation in mice. Int Immunol. 2007;19:775-84
45. Takamura K, Fukuyama S, Nagatake T, Kim DY, Kawamura A, Kawauchi H, Kiyono H. Regulatory role of lymphoid chemokine CCL19 and CCL21 in the control of allergic rhinitis. J Immunol. 2007;179:5897-906
46. Stumbles PA, Thomas JA, Pimm CL, Lee PT, Venaille TJ, Proksch S, Holt PG. Resting respiratory tract dendritic cells preferentially stimulate T helper cell type 2 (Th2) responses and require obligatory cytokine signals for induction of Th1 immunity. J Exp Med. 1998;188:2019-31
Author contact
![]() Correspondence to: David D. Lo, M.D., Ph.D., Division of Biomedical Sciences, University of California, Riverside, CA. phone: 951-827-4553, fax: 951-827-5504, email: david.loedu
Correspondence to: David D. Lo, M.D., Ph.D., Division of Biomedical Sciences, University of California, Riverside, CA. phone: 951-827-4553, fax: 951-827-5504, email: david.loedu