Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
INTRODUCTION
EPH RECEPTORS AND EPHRIN LIGANDS
EPHRIN SIGNALING PATHWAYS IN...
EPHRIN SIGNALING PATHWAYS IN...
SUMMARY
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2008; 5(5):263-272. doi:10.7150/ijms.5.263 This issue Cite
Review
Eph Receptors and Ephrin Signaling Pathways: A Role in Bone Homeostasis
Claire M. Edwards ![]() , Gregory R. Mundy
, Gregory R. Mundy
Vanderbilt Center for Bone Biology, Departments of Cancer Biology and Clinical Pharmacology/Medicine, Vanderbilt University, Nashville, TN, USA.
Received 2008-8-1; Accepted 2008-9-3; Published 2008-9-3
Abstract
The maintenance of bone homeostasis is tightly controlled, and largely dependent upon cellular communication between osteoclasts and osteoblasts, and the coupling of bone resorption to bone formation. This tight coupling is essential for the correct function and maintenance of the skeletal system, repairing microscopic skeletal damage and replacing aged bone. A range of pathologic diseases, including osteoporosis and cancer-induced bone disease, disrupt this coupling and cause subsequent alterations in bone homeostasis. Eph receptors and their associated ligands, ephrins, play critical roles in a number of cellular processes including immune regulation, neuronal development and cancer metastasis. Eph receptors are also expressed by cells found within the bone marrow microenvironment, including osteoclasts and osteoblasts, and there is increasing evidence to implicate this family of receptors in the control of normal and pathological bone remodeling.
Keywords: Bone remodeling, Eph receptors, ephrins, coupling, osteoblast, osteoclast
INTRODUCTION
The maintenance of bone homeostasis is essential for the correct function of the skeleton, including skeletal growth, repair of skeletal damage and replacement of aged bone. Bone remodeling is a continual process, and the coupling of bone resorption to bone formation is tightly controlled. The loss of this coupling and the consequent disruption of bone homeostasis is associated with a range of pathological diseases, including osteoporosis and cancer-induced bone disease. Many factors have been implicated in the control of bone homeostasis, and this review will focus on the potential role of the Eph receptor family, and the associated ephrin ligands in bone biology, both in normal and pathological conditions.
EPH RECEPTORS AND EPHRIN LIGANDS
The Eph receptors are the largest subgroup of the receptor tyrosine kinase family. They were originally identified during a screen for tyrosine kinases involved in cancer, and are named after the erythropoietin-producing hepatocellular carcinoma cell line in which the receptor was identified [1]. Eph receptors interact with ephrin ligands and there are currently 14 Eph receptors and 8 ephrin ligands identified in the human genome (http://eph-nomenclature.med.harvard.edu/). Interactions between Eph receptors and the appropriate ephrin ligand results in bi-directional signaling. Eph receptors and ephrins play a role in a number of biological processes, including cell-cell interactions, cell morphology, cell migration, angiogenesis and cancer, and there is increasing evidence for their role in normal bone homeostasis.
Structure
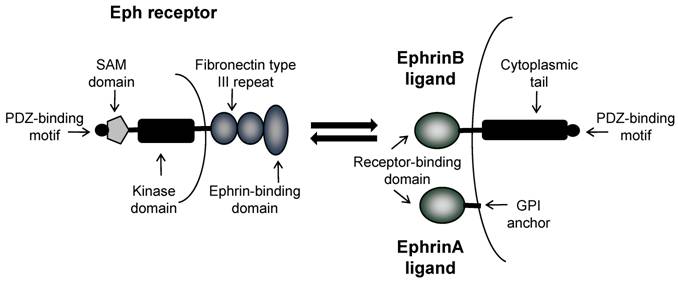
Eph receptors are divided into two classes; EphA receptors and EphB receptors; a distinction based upon their interaction with either ephrinA ligands or ephrinB ligands respectively [2]. Both EphA and EphB receptors are comprised of an extracellular region containing an ephrin-binding domain and two fibronectin type III repeats, and an intracellular region containing a juxtamembrane domain, a tyrosine kinase domain, a sterile alpha motif (SAM) and a PDZ binding domain (Figure 1). Ligand binding induces phosphorylation of the tyrosine residues within the intracellular region, resulting in a conformational change, multimerization and clustering of the Eph-ephrin complexes. EphrinA ligands are attached to the extracellular cell membrane with a glycosylphosphatidylinositol (GPI) anchor. In contrast, ephrinB ligands are transmembrane proteins containing a short cytoplasmic region. As a rule, ephrinA ligands bind EphA receptors, and ephrinB ligands bind EphB receptors, with the exception of EphA4 which can bind to ephrinA and ephrinB ligands, and ephrinA5 which can also bind to EphB2 [2, 3] .
Domain structure of Eph receptors and ephrinA and ephrinB ligands. Eph receptors have an extracellular region an ephrin-binding domain and two fibronectin type III repeats, and an intracellular region containing a tyrosine kinase domain, a SAM domain and a PDZ binding domain. EphrinA ligands are attached to the extracellular cell membrane with a GPI anchor. EphrinB ligands are transmembrane proteins with a cytoplasmic tail and PDZ binding domain. Bi-directional signaling results in forward signaling through Eph receptors and reverse signaling through ephrin ligands.
Bi-directional Signaling
An important property of interactions between Eph receptors and ephrin ligands is the bi-directional signaling that results due to activation of signaling pathways in both the receptor-expressing and the ligand-expressing cells [4]. Forward signaling is induced in the Eph receptor-expressing cells, whereas the ephrin-Eph receptor interaction also induces reverse signaling in the ephrin-expressing cell [5]. The distinct biological functions of the Eph-ephrin interaction are the result of both the multimerization of the Eph-ephrin complex and the bi-directional signaling [6].
Forward Signaling
Eph receptors are known to signal through a number of different pathways and molecules, including small GTPases of the Rho and Ras family, focal adhesion kinase (FAK), the Jak/Stat pathway and the PI3K pathway [7] [8]. Small GTPases of the Rho family mediate the effect of Eph receptor activation on actin dynamics. Rho GTPases are activated by EphA receptors, and control cell shape and movement, by promoting the formation of lamellipodia, filopodia and stress fibers [9]. This GTPase activation is mediated by exchange factors and adaptor proteins such as ephexin and Crk respectively [9] [10]. EphB receptors can also activate Rho family GTPases, mediated through the exchange factors intersectin and kalirin [11] [12]. This activation plays a role in elongation of actin filaments and morphogenesis and maturation of dendritic spines. In addition to Rho GTPases, Eph receptors can also regulate the activity of the Ras family of GTPases, including H-Ras and R-Ras [13, 14]. Activation of H-Ras leads to activation of the MAP kinase pathway, resulting in transcriptional regulation, proliferation, and cell migration. In contrast to EphA activation of Rho GTPases, the majority of Eph receptors negatively regulate the Ras-MAP kinase pathway [14]. EphB receptors can also negatively regulate the R-Ras-MAP kinase pathway, resulting in a reduction in integrin-mediated adhesion [13]. EphA receptors have also been demonstrated to regulate the Jak/Stat pathway, whereas EphB receptors promote proliferation via activation of the PI3 kinase pathway [8]. FAK is important in mediating Eph receptors and integrin signaling [7].
Reverse Signaling
The interaction between ephrin ligands and Eph receptors results not only in forward signaling through the Eph receptor, but also in 'reverse' signaling through the ephrin ligand itself [15]. Initial studies demonstrated that the extracellular domain of EphB receptors can induce tyrosine phosphorylation of ephrinB ligands [16]. A number of proteins have been identified which contain SH2 or PDZ domains, which bind to the phosphorylated ephrin ligand and transmit the signal [17, 18]. The adaptor protein, Grb4, contains an SH2 domain and is known to link ephrinB activity to cell morphology[17]. The mechanisms of reverse signaling of ephrinA ligands are less understood, but are thought to be the result of ephrinA clustering and recruitment of regulatory proteins [19].
Interactions on Same Cell Surface
Many cell types express both ephrin ligands and Eph receptors on their cell surface, raising the possibility that interactions between the ligand and receptor on the same cell may have distinct functional consequences. Evidence for the functional significance of same cell interactions was provided by studies using EphA-expressing retinal axons, which were negatively regulated by expression of ephrin A ligands on the same cell [20]. However, there is also evidence to suggest that while cells can co-express both Eph receptors and ephrin ligands, this expression is segregated into distinct membrane domains which induce opposing effects [21]. More recently, a more complex mechanism of Eph/ephrin interactions is suggested, with two distinct types of interactions identified, one of which blocks interactions which use the ligand-binding domain of the Eph receptor, and one of which uses alternative domains to inhibit EphA receptor activity [22]. Although there is still considerable work to be done to fully understand the functional significance of co-expression of Ephrin ligands and Eph receptors, evidence to date points towards an inhibitory regulatory role.
Crosstalk
In addition to the bi-directional signaling induced by Eph receptor and ephrin ligand interactions; both receptor and ligand are capable of acting independently from one another and in concert with additional non-Eph/ephrin signaling molecules. There is evidence for crosstalk between Eph receptors and the Wnt signaling pathway via Ryk, a Wnt receptor containing an inactive tyrosine kinase domain. Ryk can associate with EphB2 and EphB3, resulting in tyrosine phosphorylation [23]. EphB receptors can also directly associate with NMDA receptors at synapses [24]. Activation of EphB receptors by the ephrin ligand results in association of the Eph receptor with the NMDA receptor and promotes clustering, NMDA receptor phosphorylation and consequent calcium influx. Interactions have been reported between claudins and both EphA2 and ephrinB1, resulting in the regulation of cell adhesion [25]. Claudins have also been demonstrated to induce ephrinB1 tyrosine phosphorylation independently from Eph receptors [26]. Claudins are components of epithelial tight junctions, and are known to be expressed by bone cells including osteoblasts, therefore the potential associations between claudins and Eph/ephrins may be of functional significance in osteoblastic differentiation and bone homeostasis.
Biological Functions
Eph receptors and their ligands regulate cell-cell communication in a variety of tissues and cell types, resulting in a myriad of biological functions. They were originally identified as axon guidance molecules which mediate neuronal repulsion during CNS development, but it is now clear that their functions extend beyond that of neural development, and include critical roles in cell morphology, immune function, insulin regulation, and many aspects of cancer, including angiogenesis.
Neural Development
Eph receptors and their ligands play important roles in neural development, and are involved in both communication between individual neurons, and for communication between neurons and glial cells [27]. The bi-directional interactions regulate the regional migration of neural crest cells; during which ephrinB1 ligands have been demonstrated to both repel and promote migration [28]. EphB receptors and ephrinB ligands regulate several different aspects of synaptogenesis, including the establishment and modification of the postysynaptic specialization by transmitting signaling to the actin cytoskeleton via Rho-GTPases [24, 29]. Both EphB and Eph A receptors and ligands have been implicated in synaptic plasticity, and play a role in repair of the nervous system following injury [30-32].
Cancer
Eph receptor and ephrin ligand signaling is known to play a role in many types of cancer; indeed expression of the receptors and/or their ligands are often up-regulated in cancer cells [33]. Much of the current research points towards a tumor-suppressive role for Eph receptors, although there is also evidence for tumor-promoting effects of these receptors. The bi-directional signaling has been demonstrated to play a role in tumor angiogenesis and in tumor cell migration. In breast cancer, the most extensively studied Eph receptors are EphA2 and EphB4. Inhibition of EphB4 in breast cancer cells has been demonstrated to inhibit tumor cell survival, invasion, migration and in vivo growth [34]. Overexpression of EphA2 has been found to result in oncogenic transformation, and EphA2 kinase activity has been demonstrated to promote tumorigenesis and metastasis in murine models of breast cancer [35-37]. In contrast to this, EphA2 has also been demonstrated to have tumor suppressive effects in human breast cancer cells, highlighting the complexity of Eph receptor signaling in breast cancer. In contrast to breast cancer, in colorectal cancer, EphB receptors are thought to play a tumor suppressive role. In melanoma, increased Eph and ephrin expression correlates with metastatic progression, with evidence for roles for ephrin A1, EphA2 and ephrinB2 in both tumor suppression and progression [10, 38]. In prostate cancer and non small cell lung cancer, overexpression of EphA2 has been linked with metastasis [39, 40].
Many of the down-stream signaling targets of Eph receptors and ephrins are involved in pathways which regulate the actin cytoskeleton, as described previously. Eph receptors can also regulate integrin activity, with activation of EphA2 and EphB2 resulting in a decrease in integrin activation and cellular adhesion [7, 13]. Eph receptors can also interact with adhesion molecules such as E-cadherin to regulate cell attachment [41, 42].
Eph receptors and their ligands are known to play a role in vasculogenesis, with distinct expression of EphB4 in arterial endothelial cells and ephrinB2 in venous endothelial cells distinguishing the unique identities of these cells [43]. There is considerable evidence to support a role for Eph receptors and ephrins, from both the A and B family, in tumor angiogenesis. Forward signaling through EphA2 is known to promote angiogenesis [44]. EphA2 is expressed by tumor endothelial cells, but not during embryonic development or in quiescent adult blood vessels. The ligand ephrinB1 is expressed by both endothelial cells and tumor cells. EphA2 is required for VEGF-induced endothelial cell migration and angiogenesis [45, 46]. Stimulation by EphB4 and reverse signaling through ephrinB ligands also promotes angiogenesis [47]. EphB4 is expressed in both tumor vasculature and tumor cells, whereas ephrinB2 is expressed by tumor vasculature. The enhancement of angiogenesis through EphB4 has been demonstrated to contribute to tumor growth [47].
Immune Function
Eph receptors and their ligands are expressed in a wide range of lymphoid organs and lymphocytes [48-50]. EphB receptors have been demonstrated to regulate T cell responses and responses mediated by the T cell receptor. Of the EphB receptors, evidence is strongest to support a role of the EphB6 receptor in immune regulation, including a decreased immune response detected in EphB6 knockout mice [51]. EphA receptors and their ligands are expressed by T cells and are thought to regulate signaling through the T cell receptor [52, 53]. While expression of Eph receptors and ephrins has been detected in B lymphocytes, their function in B lymphopoiesis is unclear [50].
Insulin Regulation
The bi-directional signaling between EphA receptors and ephrinA ligands can regulate glucose homeostasis and insulin secretion [54, 55]. EphA receptors and ephrin ligands are expressed by β cells in the pancreas, and forward signaling inhibits insulin secretion, whereas reverse signaling through ephrinA ligands enhances insulin secretion. The extent of forward or reverse signaling is controlled by extracellular concentrations of glucose.
Bone Homeostasis
The maintenance of bone homeostasis is tightly controlled, and largely dependent upon cellular communication between osteoclasts and osteoblasts, and the coupling of bone resorption to bone formation. This tight coupling is essential for the correct function and maintenance of the skeletal system, repairing microscopic skeletal damage and replacing aged bone. The loss of this coupling and consequent disruption of bone homeostasis can result in a range of pathologic diseases, including osteoporosis and cancer-induced bone disease. There are many systemic and local factors which regulate both osteoclastic and osteoblastic formation and activity, for which the mechanisms of action are well described, however the communication between osteoclasts and osteoblasts during the normal process of remodeling remains poorly understood. Recent studies have implicated a role for Eph receptors and ephrin ligands in the normal coupling of bone resorption to bone formation.
Osteoclasts
Osteoclasts are large multi-nucleated terminally differentiated cells with a unique ability for bone resorption [56]. They are derived from hematopoietic stem cells, and it is the fusion of osteoclast precursor cells which results in the formation of large multi-nucleated active osteoclasts. Early differentiation of osteoclasts is dependent upon a number of transcription factors, including PU.1 [57]. The appearance of the receptor c-fms, allows the cells to undergo proliferation in response to M-CSF [58-60]. The cell is committed to the osteoclast lineage following activation of the receptor activator of nuclear factor κB (RANK) on the surface of the precursor cells, by its ligand, RANKL, which is expressed by bone marrow stromal cells and osteoblasts [61-65]. RANK activity is mediated by a number of signaling molecules, which include AP-1 transcription factors, TRAF1,2,3 5 and 6, NFATc1 and NFκB. The interaction between RANKL and RANK is critical for osteoclast formation, and can also promote osteoclast activity, since RANK is also present on the surface of terminally differentiated osteoclasts. Osteoprotegerin (OPG) is a soluble decoy receptor which can also bind to RANK, and so prevent the RANK-RANKL interaction and inhibit osteoclastogenesis. Therefore the balance of RANKL and OPG is critical for osteoclast formation and activity. There are a number of systemic factors which can indirectly regulate osteoclast formation and activity by stimulating the production of critical factors such as M-CSF and RANKL, which include PTH and IL-1. In order to resorb bone, osteoclasts attach to the bone surface via actin-rich podosomes. These enable them to form sealed zones with ruffled borders. Proteolytic enzymes such as cathepsin K, and hydrocholoric acid are secreted into this isolated environment, resulting in degradation of the bone matrix and dissolution of the bone mineral.
Osteoblasts
Osteoblasts are derived from mesenchymal stem cells, which can also differentiate into chondrocytes, fibroblasts, myocytes or adipocytes [66]. The major functions of osteoblasts are new bone formation and the regulation of osteoclastogenesis through expression of RANKL and OPG. Differentiation of mesenchymal stem cells into osteoblasts is dependent upon a number of regulatory growth factors, hormones and transcription factors. Growth factors such as bone morphogenetic protein, transforming growth factor β (TGFβ) and parathyroid hormone (PTH) play essential roles in the initial differentiation of stem cells into pre-osteoblast cells. Major transcription factors which regulate osteoblast differentiation include RUNX2, which is essential for osteoblast differentiation and plays a role in chondrocyte differentiation. The critical role of RUNX2 was identified in Runx2 null mice, which have a cartilaginous skeleton with a complete absence of osteoblasts [67, 68]. Another important transcription factor, which acts downstream of Runx2 is osterix, which is thought to direct cells away from the chondrocyte lineage towards the osteoblast lineage [69]. Following initial differentiation and proliferation, the osteoblasts stop proliferating, express alkaline phosphatase and begin to secrete collagen and non-collagenous matrix proteins such as bone sialoprotein and osteopontin. Eventually mature, mineralizing osteoblasts become embedded in the newly secreted bone matrix and undergo terminal differentiation to form osteocytes.
Bone Remodeling
Bone remodeling is a continual process which is necessary for skeletal growth and replaces damaged and aged bone [70]. The process of bone remodeling takes place in bone multicellular units throughout the skeleton. It is traditionally thought of as a cycle, comprised of activation, resorption, reversal and formation phases. The activation phase includes recruitment of osteoclast precursors. The precise cellular mechanisms responsible for osteoclast recruitment are not completely understood, but are thought to be the result of microcracks sensed by osteocytes. Hematopoietic stem cells are recruited to the site, and their differentiation to osteoclasts induced by RANKL expressed by cells of the osteoblast lineage. The osteoclasts then bind to and resorb the bone, generating a resorption lacunae during a phase which takes approximately 2-3 weeks in a human. During the reversal phase, osteoclastic bone resorption is inhibited and the osteoclasts undergo apoptosis. Osteoblasts are recruited to the site, leading to the formation phase which includes new bone formation, mineralization and subsequent quiescence.
Coupling
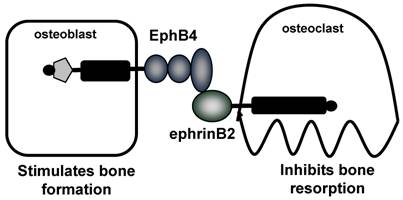
The coupling of bone resorption and bone formation is critical during the normal process of bone remodeling, and the dysregulation of this coupling results in the development of a range of pathological bone diseases. There is considerable evidence to support the coupling of bone formation to bone resorption, however the mechanisms responsible are unclear. It is known that in vivo, stimulation of bone resorption is accompanied by an increase in bone formation, and it is these studies which led to the idea of a locally produced 'coupling factor' [71]. Several studies have implicated growth factors, including IGF-I and II and TGF-β, which are released from the bone matrix during bone resorption and can stimulate osteoblast differentiation [72, 73]. Another potential mechanism is that the coupling factor is released from the osteoclasts, upon inhibition of resorptive activity [74]. Evidence for this theory comes from genetic mouse models, including mice where the SHP-ras-MAPK pathway was inactivated, resulting in an increase in osteoclasts, bone resorption and bone formation, which was thought to be dependent upon active osteoclasts and IL-6 . [75] In addition, OPG deficient mice were found to have not only an increase in osteoclast formation, but also an increase in bone formation which was thought to be the result of cellular factors [76]. Calcitonin deficient mice also support the notion that the activated osteoclast is important for coupling. Calcitonin is well known to inhibit osteoclast function, however these mice display an increase in bone formation, an effect postulated to be the result of continuous osteoclast activation due to the calcitonin deficiency [77]. In vitro studies have implicated several factors secreted from osteoclasts, which have been found to have direct effects on osteoblasts to promote differentiation, including sphingosine 1-phosphate (S1P), myb-induced myeloid protein-1 (mim-1), and hepatocyte growth factor (HGF) [78-80]. More recently, as will be discussed, a new concept for the coupling of bone resorption to bone formation has been proposed, involving bidirectional signaling between EphB4 receptor on osteoblasts and ephrinB2 on osteoclasts [81]. The cellular and molecular mechanisms responsible for the coupling of bone resorption to bone formation must be able to explain the unique properties of this process. For example, (i) the localized nature of coupling, which starts with resorption and is followed by bone formation, occurring only at sites of prior resorption, and (ii) the cessation of bone resorption upon commencement of bone formation. These suggest both local mechanism, and the necessity for signaling to both osteoblasts to stimulate formation and to osteoclasts to inhibit formation, for which bi-directional signaling between osteoblasts and osteoclasts provides a novel and intriguing potential explanation.
Proposed coupling of bone resorption and bone formation via EphB4 and ephrinB2. Zhao and collegues demonstrate expression of EphB4 on osteoblasts and ephrinB2 on osteoclasts. Forward signaling through EphB4 stimulates bone formation, whereas reverse signaling through ephrinB2 inhibits bone resorption [81]. Therefore, the interaction between EphB4 and ephrinB2 results in a switch from resorption to formation.
EPHRIN SIGNALING PATHWAYS IN BONE BIOLOGY
It is only in recent years that a potential role of Eph receptors and ephrins in bone biology has emerged. At present, there is strong evidence to suggest a role for the ephrinB/EphB family in bone biology. With the exception of a role in cancer bone metastasis the role of the ephrinA/EphA family has not been investigated.
EphrinB1
The role of ephrinB1 in skeletal development was first investigated by Compagni et al., who used Cre-lox technology to create an ephrinB1 knockout mouse [82]. The global deletion of EphrinB1 resulted in perinatal lethality, edema, defective body wall closure and skeletal abnormalities. The skeletal abnormalities affected both the axial and appendicular skeleton and included cleft palate, shortening of the skull, asymmetric paring of the ribs, sternebral fusions and polydactyly affecting digits I or II. The asymptomatic pairing of the ribs and sternebral fusions were also seen in EphB2/EphB3 double knockout mice, indicating the importance of ephrinB1-EphB4 interactions in rib development. Furthermore, the skeletal defects associated with the ephrinB1 phenotype were only reproduced in double knockout mice, lacking both EphB2 and EphB3, indicating a degree of functional redundancy in these receptors. Preaxial polydactyly was exclusively seen in heterozygous females in which expression of the X-linked ephrinB1 gene was mosaic. The ectopic EphB-ephrinB1 interactions at mosaic interfaces were sufficient to induce splitting of chondrogenic condensations by generating restricting cell movement. To further examine the mechanisms behind the limb defects in ephrinB1 knockout mice, Compagni et al. utilized the Prx-Cre transgenic mouse to create a limb-specific ephrinB1 knockout, in which the preaxial polydactyly was still present. Despite evidence for the involvement of the sonic hedgehog pathway in polydactyly, no evidence was found for a role for this pathway in the polydactyly observed in the ephrinB1 knockout mice. Defects were also detected in the wrist skeleton, including the fusion of distal carpal bones and the formation of ectopic ossifications. EphrinB1 protein was observed in prechondrogenic condensations, and the receptors EphB2 and EphB3 were found on adjacent mesenchymal stem cells.
In support of these observations, Davy and colleagues have also observed perinatal lethality and skeletal defects in ephrinB1 deficient mice [83]. Limb bud cultures from wildtype and ephrinB1 knockout mice suggested that the role of ephrinB1 in digit formation may involve perichondrium formation or maintenance. In addition to generating global ephrinB1 knockout mice, they also generated mice with a mutation in ephrinB1 in which the PDZ binding domain was mutated. The PDZ binding domain is necessary for reverse signaling through ephrinB1, and mutating this specific domain revealed a cell autonomous role for ephrinB1 in neural crest cells. Targeted disruption of ephrinB1 was found to reduce bone size in vivo. EphrinB1 was targeted to cells in the mesenchymal lineage, including osteoblasts, using the Col1a2 promoter and this inhibition was found to decrease peak bone mass and bone size [84].
Mutations in the ephrinB1 gene have been associated with craniofrontonasal syndrome in humans [85, 86]. Craniofrontonasal syndrome (CFNS) is an X-linked developmental disorder in which affected females exhibit multiple skeletal malformations, including asymmetry of craniofacial structures and abonormalities of the thoracic skeleton. A gene for CFNS has been mapped to the pericentromeric region of the X chromosome, and the ephrinB1 gene is localized within this mapping interval [87]. The analysis of three families with CFNS revealed a deletion of exons 2-5 of ephrinB1 gene in one family, and missense mutations resulting in amino acid exchanges in two families [85]. The mutations were located in multimerization and receptor-interaction motifs within the ephrinB1 extracellular domain. In all cases, mutations were found in male carriers, clinically affected males, and affected heterozygous females. In a separate study, Twigg and colleagues identified mutations in 24 females with CFNS, from 20 different families [86]. The location of these mutations suggest that they would result in complete or partial loss of EphrinB1 function. The ephrinB1 gene is X-inactivated, however there was no indication of markedly skewed X-inactivation in either blood or cranial periosteum from females with CFNS, indicating that the lack of ephrinB1 does not compromise cell viability. The authors propose that the fusion of the coronal sutures associated with females with CFNS is due to a patchwork loss of ephinB1 expression resulting in disturbance at the tissue boundary formation of the developing coronal suture. These studies confirm the involvement of ephrinB1 in human skeletal development.
EphrinB2
The initial identification of a potential role for ephrinB2 in bone biology came from the discovery that ephrinB2 was a target gene of NFAT that was upregulated during osteoclast differentiation [81]. EphrinB2 protein was induced during osteoclast differentiation, and detected in both multinucleated osteoclasts and differentiating mononuclear osteoclasts. Osteoclasts were not found to express the corresponding EphB receptors, however osteoblasts were found to constitutively express both ephrin ligands and Eph receptors. Reverse signaling through ephrinB2 on osteoclasts was found to suppress osteoclast formation. The intracellular domain of ephrinB2 was found to be essential for reverse signaling, and the inhibitory signals were found to be dependent upon interactions with the PDZ domain, and inhibition of Fos and NfatC1 transcription, but not dependent upon tyrosine phosphorylation. Despite strong in vitro evidence that ephrinB2 can inhibit osteoclastogenesis, mice lacking ephrinB2 in macrophages and osteoclasts were not found to have a significant bone phenotype, an effect attributed to compensation by ephrinB1. Although ephrinB2 can interact with all EphB receptors, only EphB4 can stimulate reverse signaling through ephrinB2. Therefore, the authors investigated the ephrinB2-EphB4 interactions, with a focus on the role of EphB4 in osteoblasts. EphrinB2 was found to stimulate forward signaling through EphB4, resulting in an increase in osteoblast formation, potentially mediated by RhoA inactivation. Support for a role for EphB4 in osteoblast biology was provided by EphB4 transgenic mice, where EphB4 overexpression was directed to cells of the osteoblast lineage using the Col1a1 promoter. These mice demonstrated an increase in bone mass, bone mineral density and bone formation rates. Furthermore, osteoclast number was decreased, suggesting that EphB4 overexpression also inhibited osteoclast function. No changes in RANKL or OPG were detected. Taken together, these results suggest that increased EphB4 expression in osteoblasts enhances bone formation and inhibits bone resorption in vivo.
In addition to the forward and reverse signaling induced by ephrinB2 expressed on osteoclasts, there is also evidence for a role for ephrinB2 expressed on osteoblasts in osteoblast differentiation and bone formation [88]. EphrinB2 expression was found to be increased on a mouse bone marrow stromal cell line in response to treatment with both PTH and PTHrP, and in vivo osteoblastic expression was confirmed in mouse femurs by immunohistochemistry. Expression of ephrinB2 was not altered during osteoblast differentiation. Allan et al. used a specific peptide inhibitor of ephrinB2/EphB4 to determine the effect of interactions between ephrinB2 and EphB4 in osteoblasts; demonstrating a significant inhibition of mineralization. These results demonstrate the potential for autocrine or paracrine effects of osteoblastic ephrinB2 on EphB4 in osteoblasts, and suggest that these effects may contribute to the anabolic effect of PTH or PTHrP. Further evidence for a role for ephrinB2 in osteoblasts is provided by Wang et al., who determined that inhibition of IGF-1R in osteoblasts decreased ephrinB2 expression and prevented the PTH-induced increase in ephrinB2, thus implicating IGF-1R in mediating the effects of PTH on ephrinB2 and ephrinB4 [89]. Furthermore, Xing et al., identified ephrinB2 as one of a number of genes that was differentially expressed in mouse tibia following mechanical loading [90].
EPHRIN SIGNALING PATHWAYS IN CANCER-INDUCED BONE DISEASE
The increasing evidence for a role for ephrin and Eph receptor signaling in bone biology raises the possibility that these receptor/ligand interactions may be important in diseases with dysregulated bone remodeling. Breast cancer bone metastases are associated with the development of an osteolytic bone disease, and a recent study has implicated EphA2 as a potential mediator of this bone destruction [91]. Overexpression of a truncated mutant of EphA2 in breast cancer cells was found to inhibit the development of osteolytic bone lesions in vivo. This suggests that expression of EphA2 by breast cancer cells may promote the development of osteolytic bone disease. Multiple myeloma is associated with an osteolytic bone disease characterized by an increase in osteoclastic bone resorption and a reduction in bone formation. The cellular and molecular mechanisms which mediate the uncoupling of bone resorption from bone formation in myeloma are poorly understood. Our own studies have demonstrated that myeloma cells can down-regulate EphB4 expression in osteoblasts, suggesting that the reduction in bone formation in myeloma bone disease is mediated by a reduction in EphB4 expression and thus disruption of the normal coupling of bone resorption and bone formation [92]. Bone is a frequent site of metastasis for prostate cancer, and tissue microarray analysis of metastatic foci in lymph nodes, liver and bone identified decreased expression of ephrinA1 specifically in bone metastases [93]. Giant cell tumors of bone are primary bone tumors associated with osteolysis. Microarray analysis comparing primary and recurrent giant cell tumors determined that EphA1 expression was decreased in the recurrent tumors [94]. This decreased expression was confirmed at the protein level by immunohistochemistry, implicating EphA1 in the progression of giant cell tumors of bone.
SUMMARY
The Eph receptor family and the associated ephrin ligands play critical roles in many cellular processes, and the complexity of the bidirectional signaling increases the functions of the ligand-receptor interaction. Their role in neural development and angiogenesis is well documented, however their potential role in bone biology is only now beginning to emerge. Despite many significant advances in bone biology, many questions remain unanswered, including that of the nature of the 'coupling' of bone resorption to bone formation. The potential role of Eph receptors and ephrin ligands in this coupling is intriguing, suggesting a new concept for coupling and osteoblast-osteoclast communication. Furthermore, the increasing evidence for a role for Eph receptors and their ligands in cancer-associated bone disease identifies new molecular pathways and potentially novel therapeutic targets for the treatment of these destructive and, for the most part, fatal diseases. Many questions remain still to be answered, including the cellular and molecular consequences of the bidirectional signaling in bone biology and the function of the additional members of this large receptor family, in order to fully determine the role of the Eph receptors and ephrin ligands in bone homeostasis.
CONFLICT OF INTERESTS
The authors have declared that no conflict of interest exists.
References
1. Hirai H, Maru Y, Hagiwara K. et al. A novel putative tyrosine kinase receptor encoded by the eph gene. Science. 1987;238:1717-1720
2. Kullander K, Klein R. Mechanisms and functions of eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475-486
3. Himanen JP, Chumley MJ, Lackmann M. et al. Repelling class discrimination: Ephrin-a5 binds to and activates ephb2 receptor signaling. Nat Neurosci. 2004;7:501-509
4. Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462-475
5. Egea J, Klein R. Bidirectional eph-ephrin signaling during axon guidance. Trends Cell Biol. 2007;17:230-238
6. Stein E, Lane AA, Cerretti DP. et al. Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genes Dev. 1998;12:667-678
7. Miao H, Burnett E, Kinch M. et al. Activation of epha2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62-69
8. Lai KO, Chen Y, Po HM. et al. Identification of the jak/stat proteins as novel downstream targets of epha4 signaling in muscle: Implications in the regulation of acetylcholinesterase expression. Biol Chem J. 2004;279:13383-13392
9. Shamah SM, Lin MZ, Goldberg JL. et al. Epha receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105:233-244
10. Lawrenson ID, Wimmer-Kleikamp SH, Lock P. et al. Ephrin-a5 induces rounding, blebbing and de-adhesion of epha3-expressing 293t and melanoma cells by crkii and rho-mediated signalling. Cell Sci J. 2002;115:1059-1072
11. Irie F, Yamaguchi Y. Ephb receptors regulate dendritic spine development via intersectin, cdc42 and n-wasp. Nat Neurosci. 2002;5:1117-1118
12. Penzes P, Beeser A, Chernoff J. et al. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinb-ephb receptor activation of the rho-gef kalirin. Neuron. 2003;37:263-274
13. Zou JX, Wang B, Kalo MS. et al. An eph receptor regulates integrin activity through r-ras. Proc Natl Acad Sci U S A. 1999;96:13813-13818
14. Miao H, Wei BR, Peehl DM. et al. Activation of epha receptor tyrosine kinase inhibits the ras/mapk pathway. Nat Cell Biol. 2001;3:527-530
15. Bruckner K, Pasquale EB, Klein R. Tyrosine phosphorylation of transmembrane ligands for eph receptors. Science. 1997;275:1640-1643
16. Holland SJ, Gale NW, Mbamalu G. et al. Bidirectional signalling through the eph-family receptor nuk and its transmembrane ligands. Nature. 1996;383:722-725
17. Cowan CA, Henkemeyer M. The sh2/sh3 adaptor grb4 transduces b-ephrin reverse signals. Nature. 2001;413:174-179
18. Lu Q, Sun EE, Klein RS, Flanagan JG. Ephrin-b reverse signaling is mediated by a novel pdz-rgs protein and selectively inhibits g protein-coupled chemoattraction. Cell. 2001;105:69-79
19. Davy A, Gale NW, Murray EW. et al. Compartmentalized signaling by gpi-anchored ephrin-a5 requires the fyn tyrosine kinase to regulate cellular adhesion. Genes Dev. 1999;13:3125-3135
20. Hornberger MR, Dutting D, Ciossek T. et al. Modulation of epha receptor function by coexpressed ephrina ligands on retinal ganglion cell axons. Neuron. 1999;22:731-742
21. Marquardt T, Shirasaki R, Ghosh S. et al. Coexpressed epha receptors and ephrin-a ligands mediate opposing actions on growth cone navigation from distinct membrane domains. Cell. 2005;121:127-139
22. Carvalho RF, Beutler M, Marler KJ. et al. Silencing of epha3 through a cis interaction with ephrina5. Nat Neurosci. 2006;9:322-330
23. Schmitt AM, Shi J, Wolf AM. et al. Wnt-ryk signalling mediates medial-lateral retinotectal topographic mapping. Nature. 2006;439:31-37
24. Dalva MB, Takasu MA, Lin MZ. et al. Ephb receptors interact with nmda receptors and regulate excitatory synapse formation. Cell. 2000;103:945-956
25. Tanaka M, Kamata R, Sakai R. Epha2 phosphorylates the cytoplasmic tail of claudin-4 and mediates paracellular permeability. Biol Chem J. 2005;280:42375-42382
26. Tanaka M, Kamata R, Sakai R. Phosphorylation of ephrin-b1 via the interaction with claudin following cell-cell contact formation. Embo J. 2005;24:3700-3711
27. Yamaguchi Y, Pasquale EB. Eph receptors in the adult brain. Curr Opin Neurobiol. 2004;14:288-296
28. Santiago A, Erickson CA. Ephrin-b ligands play a dual role in the control of neural crest cell migration. Development. 2002;129:3621-3632
29. Ethell IM, Irie F, Kalo MS. et al. Ephb/syndecan-2 signaling in dendritic spine morphogenesis. Neuron. 2001;31:1001-1013
30. Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: Signalling functions at the synapse. Nat Rev Neurosci. 2007;8:206-220
31. Du J, Fu C, Sretavan DW. Eph/ephrin signaling as a potential therapeutic target after central nervous system injury. Curr Pharm Des. 2007;13:2507-2518
32. Liu X, Hawkes E, Ishimaru T. et al. Ephb3: An endogenous mediator of adult axonal plasticity and regrowth after cns injury. Neurosci J. 2006;26:3087-3101
33. Brantley-Sieders D, Schmidt S, Parker M, Chen J. Eph receptor tyrosine kinases in tumor and tumor microenvironment. Curr Pharm Des. 2004;10:3431-3442
34. Kumar SR, Singh J, Xia G. et al. Receptor tyrosine kinase ephb4 is a survival factor in breast cancer. Am Pathol J. 2006;169:279-293
35. Zelinski DP, Zantek ND, Stewart JC. et al. Epha2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61:2301-2306
36. Brantley-Sieders DM, Fang WB, Hicks DJ. et al. Impaired tumor microenvironment in epha2-deficient mice inhibits tumor angiogenesis and metastatic progression. Faseb J. 2005;19:1884-1886
37. Fang WB, Brantley-Sieders DM, Parker MA. et al. A kinase-dependent role for epha2 receptor in promoting tumor growth and metastasis. Oncogene. 2005;24:7859-7868
38. Easty DJ, Hill SP, Hsu MY. et al. Up-regulation of ephrin-a1 during melanoma progression. Int Cancer J. 1999;84:494-501
39. Walker-Daniels J, Coffman K, Azimi M. et al. Overexpression of the epha2 tyrosine kinase in prostate cancer. Prostate. 1999;41:275-280
40. Kinch MS, Moore MB, Harpole DH. Predictive value of the epha2 receptor tyrosine kinase in lung cancer recurrence and survival. Clin Cancer Res. 2003;9:613-618
41. Orsulic S, Kemler R. Expression of eph receptors and ephrins is differentially regulated by e-cadherin. Cell Sci J. 2000;113(Pt 10):1793-1802
42. Zantek ND, Azimi M, Fedor-Chaiken M. et al. E-cadherin regulates the function of the epha2 receptor tyrosine kinase. Cell Growth Differ. 1999;10:629-638
43. Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-b2 and its receptor eph-b4. Cell. 1998;93:741-753
44. Brantley DM, Cheng N, Thompson EJ. et al. Soluble eph a receptors inhibit tumor angiogenesis and progression in vivo. Oncogene. 2002;21:7011-7026
45. Cheng N, Brantley D, Fang WB. et al. Inhibition of vegf-dependent multistage carcinogenesis by soluble epha receptors. Neoplasia. 2003;5:445-456
46. Cheng N, Brantley DM, Liu H. et al. Blockade of epha receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol Cancer Res. 2002;1:2-11
47. Noren NK, Lu M, Freeman AL. et al. Interplay between ephb4 on tumor cells and vascular ephrin-b2 regulates tumor growth. Proc Natl Acad Sci U S A. 2004;101:5583-5588
48. Munoz JJ, Alonso CL, Sacedon R. et al. Expression and function of the eph a receptors and their ligands ephrins a in the rat thymus. Immunol J. 2002;169:177-184
49. Wu J, Luo H. Recent advances on t-cell regulation by receptor tyrosine kinases. Curr Opin Hematol. 2005;12:292-297
50. Aasheim HC, Munthe E, Funderud S. et al. A splice variant of human ephrin-a4 encodes a soluble molecule that is secreted by activated human b lymphocytes. Blood. 2000;95:221-230
51. Luo H, Yu G, Tremblay J, Wu J. Ephb6-null mutation results in compromised t cell function. Clin Invest J. 2004;114:1762-1773
52. Freywald A, Sharfe N, Miller CD. et al. Epha receptors inhibit anti-cd3-induced apoptosis in thymocytes. Immunol J. 2006;176:4066-4074
53. Sharfe N, Nikolic M, Cimpeon L. et al. Epha and ephrin-a proteins regulate integrin-mediated t lymphocyte interactions. Mol Immunol. 2008;45:1208-1220
54. Konstantinova I, Nikolova G, Ohara-Imaizumi M. et al. Epha-ephrin-a-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell. 2007;129:359-370
55. Kulkarni Kahn CR RN. Ephs and ephrins keep pancreatic beta cells connected. Cell. 2007;129:241-243
56. Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638-649
57. Tondravi MM, McKercher SR, Anderson K. et al. Osteopetrosis in mice lacking haematopoietic transcription factor pu.1. Nature. 1997;386:81-84
58. Felix R, Cecchini MG, Fleisch H. Macrophage colony stimulating factor restores in vivo bone resorption in the op/op osteopetrotic mouse. Endocrinology. 1990;127:2592-2594
59. Kodama H, Nose M, Niida S, Yamasaki A. Essential role of macrophage colony-stimulating factor in the osteoclast differentiation supported by stromal cells. Exp Med J. 1991;173:1291-1294
60. Yoshida H, Hayashi S, Kunisada T. et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442-444
61. Y-Kong Y, Yoshida H, Sarosi I. et al. Opgl is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315-323
62. Lacey DL, Timms E, Tan HL. et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165-176
63. Li J, Sarosi I, Yan XQ. et al. Rank is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A. 2000;97:1566-1571
64. Mizuno A, Amizuka N, Irie K. et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochemical and Biophysical Research Communications. 1998;247:610-615
65. Simonet WS, Lacey DL, Dunstan CR. et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309-319
66. Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349-355
67. Ducy P, Zhang R, Geoffroy V. et al. Osf2/cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89:747-754
68. Komori T, Yagi H, Nomura S. et al. Targeted disruption of cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755-764
69. Nakashima K, Zhou X, Kunkel G. et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17-29
70. Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791-801
71. Howard GA, Bottemiller BL, Turner RT. et al. Parathyroid hormone stimulates bone formation and resorption in organ culture: Evidence for a coupling mechanism. Proc Natl Acad Sci U S A. 1981;78:3204-3208
72. Hayden JM, Mohan S, Baylink DJ. The insulin-like growth factor system and the coupling of formation to resorption. Bone. 1995;17:93S-98S
73. Centrella M, McCarthy TL, Canalis E. Transforming growth factor-beta and remodeling of bone. J Bone Joint Surg Am. 1991;73:1418-1428
74. Parfitt AM, Mundy GR, Roodman GD. et al. A new model for the regulation of bone resorption, with particular reference to the effects of bisphosphonates. J Bone Miner Res. 1996;11:150-159
75. Sims NA, Jenkins BJ, Quinn JM. et al. Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. Clin Invest J. 2004;113:379-389
76. Nakamura M, Udagawa N, Matsuura S. et al. Osteoprotegerin regulates bone formation through a coupling mechanism with bone resorption. Endocrinology. 2003;144:5441-5449
77. Hoff AO, Catala-Lehnen P, Thomas PM. et al. Increased bone mass is an unexpected phenotype associated with deletion of the calcitonin gene. Clin Invest J. 2002;110:1849-1857
78. Ryu J, Kim HJ, Chang EJ. et al. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. Embo J. 2006;25:5840-5851
79. Falany ML, AM Thames 3rd, McDonald JM. et al. Osteoclasts secrete the chemotactic cytokine mim-1. Biochem Biophys Res Commun. 2001;281:180-185
80. Grano M, Galimi F, Zambonin G. et al. Hepatocyte growth factor is a coupling factor for osteoclasts and osteoblasts in vitro. Proceedings of the National Acadamy of Sciences of the USA. 1996;93:7644-7648
81. Zhao C, Irie N, Takada Y. et al. Bidirectional ephrinb2-ephb4 signaling controls bone homeostasis. Cell Metab. 2006;4:111-121
82. Compagni A, Logan M, Klein R, Adams RH. Control of skeletal patterning by ephrinb1-ephb interactions. Dev Cell. 2003;5:217-230
83. Davy A, Aubin J, Soriano P. Ephrin-b1 forward and reverse signaling are required during mouse development. Genes Dev. 2004;18:572-583
84. Xing W, Govoni K, Kapoor A. et al. Targeted disruption of ephrin b1 in osteoblasts reduces bone size in mice. J Bone Miner Res. 2007;22:1107
85. Wieland I, Jakubiczka S, Muschke P. et al. Mutations of the ephrin-b1 gene cause craniofrontonasal syndrome. Am Hum Genet J. 2004;74:1209-1215
86. Twigg SR, Kan R, Babbs C. et al. Mutations of ephrin-b1 (efnb1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci U S A. 2004;101:8652-8657
87. Wieland I, Jakubiczka S, Muschke P. et al. Mapping of a further locus for x-linked craniofrontonasal syndrome. Cytogenet Genome Res. 2002;99:285-288
88. Allan EH, Hausler KD, Wei T. et al. Ephrinb2 regulation by parathyroid hormone (pth) and pthrp revealed by molecular profiling in differentiating osteoblasts. J Bone Miner Res. 2008 [Epub ahead of print]
89. Wang Y, ElAlieh HZ, Chang W. et al. Ablation of igf-1 signaling disrupts the communication between osteoblasts and osteoclasts. J Bone Miner Res. 2007;22:S241
90. Xing W, Baylink D, Kesavan C. et al. Global gene expression analysis in the bones reveals involvement of several novel genes and pathways in mediating an anabolic response of mechanical loading in mice. Cell Biochem J. 2005;96:1049-1060
91. Vaught D, Brantley-Sieders D, Chen J. Epha2 induced osteolysis: A novel mechanism for osteoclast activation mediated by breast cancer-bone cell interactions. Proceedings of the 9th Annual Meeting of the American Association for Cancer Research. 2008: 2523.
92. Bates AL, Mundy GR, Edwards CM. Myeloma cells decrease ephb4 expression in osteoblasts: A novel mechanism for regulation of bone formation in multiple myeloma. J Bone Miner Res. 2007;22:S309
93. Morrissey C, True LD, Roudier MP. et al. Differential expression of angiogenesis associated genes in prostate cancer bone, liver and lymph node metastases. Clin Exp Metastasis. 2008;25:377-388
94. Guenther R, Krenn V, Morawietz L. et al. Giant cell tumors of the bone: Molecular profiling and expression analysis of ephrin a1 receptor, claudin 7, cd52, fgfr3 and amfr. Pathol Res Pract. 2005;201:649-663
Author contact
![]() Correspondence to: Claire M. Edwards, Vanderbilt Center for Bone Biology, 2215 Garland Avenue, Room 1235, Vanderbilt University, Nashville, TN 37232-0575. Phone: 615 343 2801; Fax: 615 343 2611; Email: claire.edwardsedu
Correspondence to: Claire M. Edwards, Vanderbilt Center for Bone Biology, 2215 Garland Avenue, Room 1235, Vanderbilt University, Nashville, TN 37232-0575. Phone: 615 343 2801; Fax: 615 343 2611; Email: claire.edwardsedu