Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2008; 5(1):1-8. doi:10.7150/ijms.5.1 This issue Cite
Research Paper
Differential Constitutive and Cytokine-Modulated Expression of Human Toll-like Receptors in Primary Neutrophils, Monocytes, and Macrophages
D. Shane O'Mahony1, Uyenvy Pham1, Ramesh Iyer1, Thomas R. Hawn1, W. Conrad Liles1,2,3 ![]()
1. Departments of Medicine, University of Washington, Seattle, WA 98195, USA
2. Departments of Pathology, University of Washington, Seattle, WA 98195, USA
3. McLaughlin-Rotman Centre for Global Health, Toronto General Research Institute, University Health Network, and Department of Medicine, University of Toronto, Toronto, ON M5G 2C4, CANADA
Received 2007-11-14; Accepted 2008-1-3; Published 2008-1-4
Abstract
Human Toll-like receptors (TLRs) comprise a family of proteins that recognizes pathogen-associated molecular patterns (PAMPs) and initiates host innate immune responses. Neutrophils, monocytes, and macrophages are critical cellular components of the human innate immune system. Proinflammatory cytokines, such as granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage colony-stimulating factor (M-CSF), and interferon-γ (IFN-γ), have been shown to up-regulate microbicidal activity in these effector cells of innate immunity. Currently, the cellular and molecular mechanisms responsible for these effects are not completely understood. We hypothesized that these cytokines may up-regulate TLR expression as a mechanism to facilitate microbial recognition and augment the innate immune response. Using quantitative realtime rt-PCR technology, we examined constitutive expression of TLR2, TLR4, TLR5, and TLR9 mRNA and the effects of G-CSF, GM-CSF, M-CSF, and IFN-γ on TLR mRNA expression in purified populations of normal human neutrophils, monocytes, and monocyte-derived macrophages. Relative constitutive expression of TLR2, TLR4, and TLR9 was similar in neutrophils and monocytes. Constitutive expression of TLR5 was less in neutrophils compared to monocytes. Constitutive expression of TLR4 was greater and that of TLR9 lower in monocyte-derived macrophages compared to monocytes. Of the cytokines examined, IFN-γ and GM-CSF caused the greatest effects on TLR expression. IFN- γ up-regulated TLR2 and TLR4 in neutrophils and monocytes. GM-CSF up-regulated expression of TLR2 and TLR4 in neutrophils and TLR2 in monocytes. TLR5 was down-regulated by inflammatory cytokines in monocytes. These results suggest a potential role for IFN- γ and/or GM-CSF as therapeutic immunomodulators of the host defense to infection.
Keywords: Toll-like receptor, human, phagocytes, cytokines, innate immunity
Introduction
The professional phagocytes, namely neutrophils, monocytes and macrophages, play important roles in host defense as critical cellular components of the innate immune system. These leukocytes have the ability to quickly recognize pathogens, mediate phagocytosis, and respond with production of reactive oxygen and nitrogen species, antimicrobial peptides, and inflammatory mediators.[1] The rapid action of the innate immune system depends on microbial product recognition conferred by germline-encoded pattern recognition receptors (PRRs), including CD14, β2-integrins, C-type lectins, macrophage scavenger receptors, complement receptors, and the recently identified members of the mammalian Toll-like receptor (TLRs).[2] To date, at least 10 human TLRs have been identified through DNA sequencing.[3-9] TLRs have specificity for highly conserved pathogen-associated molecular patterns (PAMPs) and may interact with other TLRs and/or other PRRs in recognition and subsequent intracellular signaling, thereby enabling the innate immune system to respond to pathogens discriminately.[10-12] After ligand interaction with TLRs, intracellular signaling occurs via adaptor proteins, including MyD88, TIRAP/Mal, TRIF and TRAM, which induce a downstream signaling cascade that culminates in nuclear translocation of NF-κB.[13-15]
The expression patterns of TLRs in different cell types may be an important regulatory mechanism of the innate immune response to various pathogens. Flow cytometry studies have demonstrated constitutive TLR expression in various tissues, especially leukocytes.[16-18] TLR1, TLR2, TLR4, TLR5, and TLR6 are expressed on the cell surface, as well as intracellularly where they can be recruited to phagolysosomes. [11, 18] Expression of TLR3, TLR7, TLR8, and TLR9 is localized predominantly to intracellular compartments where they recognize viral and/or bacterial nucleic acids.[19-21] Neutrophils constitutively express all TLRs except TLR3, whereas monocytes appear to lack expression of TLR3, TLR6, TLR7, and TLR10. [18, 22, 23] TLR3 expression is limited to endosomes of myeloid and monocyte-derived dendritic cells (DCs). Therefore, myeloid DCs are the only identified cell type which express the entire repertoire of TLRs. On the other hand, plasmacytoid DCs have more limited expression patterns. [22, 24] Recently, Peng et al have shown that regulatory T-cells express TLR8 and that the TLR8-MyD88 signaling pathway controls suppressive function of Treg cells.[25] These findings suggest that the unique expression patterns of TLRs in various tissues are important for their functional biologic role in immunity.
TLR2, TLR4, TLR5 and TLR9 play critical roles in innate immune recognition of bacteria (Reviewed in [15]). TLR1 and TLR6 contribute to discrimination of bacterial lipopeptides by functionally combining with TLR2.[26] TLR2 has been associated with recognition of cell wall structures of gram-positive bacilli, mycoplasma, mycobacteria and yeast including lipoproteins, glycolipids, and zymosan.[27-32] TLR4 is considered fundamental to detection of LPS from gram-negative bacteria.[33, 34] Bacterial flagellin has been shown to be the ligand for TLR5, and TLR9 recognizes CpG motifs on bacterial DNA.[21, 35, 36] Bacterial products and proinflammatory cytokines have been shown to contribute to the regulation of TLR expression on monocytes and neutrophils, but the role of pro-inflammatory cytokines in regulation of TLR expression remains unclear and discrepant in previous reports.[16, 17, 22, 37-40]
Pro-inflammatory cytokines, such as granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage colony-stimulating factor (M-CSF), and interferon-γ (IFN-γ), have been shown to up-regulate microbicidal activity of neutrophils and/or monocytes/macrophages. All four of these cytokines, which are available in recombinant human form, have been proposed and considered as adjunctive immunomodulatory agents to treat serious or refractory infections in humans.[41, 42] Currently, the precise cellular and molecular mechanisms responsible for these effects are not completely understood but may potentially include up-regulation of molecules involved in pathogen recognition, such as TLRs. Indeed, recent studies demonstrated that pretreatment of neutrophils with GM-CSF modulates TLR expression and cytokine responses to several TLR ligands.[37, 38]
In this study, we systematically investigated the effects of G-CSF, GM-CSF, M-CSF, and IFN-γ on TLR mRNA expression in human leukocytes, focusing on the TLRs involved in bacterial recognition. Monocytes and neutrophils isolated from normal human blood were cultured in the absence and presence of these cytokines, and realtime rt-PCR technology was employed to quantitatively measure relative transcription of TLR2, TLR4, TLR5 and TLR9.
Methods
Isolation and culture of cells
Human monocytes and neutrophils were isolated from EDTA-anticoagulated peripheral venous blood of normal human volunteers according to a protocol approved in advance by the Human Subjects Committee of the University of Washington Investigational Review Board. Monocytes were isolated by negative immunoselection with RosetteSep Human Monocyte Enrichment Cocktail as described by the manufacturer (Stem Cell Technologies, Vancouver, Canada). Neutrophils were isolated from EDTA-anticoagulated blood by sequential sedimentation in dextran (Sigma, St. Louis, MO) in 0.9% sodium chloride, centrifugation over Histopaque-1077 (Sigma), and hypotonic lysis of erythrocytes, as previously described.[43] Cells were cultured in RPMI 1640 (BioWhittaker, Walkersville, MD) supplemented with 10% heat-inactivated fetal bovine serum (BioWhittaker), 1% L-glutamine, 1% HEPES, and 0.5% penicillin-streptomycin (all from BioWhittaker). Isolated monocytes were used immediately or were incubated at 37˚C in a 5% CO2-controlled incubator for the designated time period without stimuli or in the presence of recombinant human IFN-γ (1000 units/ml) (R&D Systems Minneapolis, Minnesota), recombinant human GM-CSF (100 ng/ml) (Immunex, Seattle, WA), recombinant human M-CSF (100 ng/ml) (R&D Systems), or LPS (1 μg/ml) (Sigma). Neutrophils were used immediately or were incubated with recombinant human IFN- γ (1000 units/ml), recombinant human G-CSF (100 ng/ml) (Amgen, Thousand Oaks, CA), recombinant human GM-CSF (100 ng/ml), or LPS (1 μg/ml). The doses of cytokines were chosen based on previous work in our lab that demonstrated what the doses yielded maximal activation of human neutrophils and monocytes/macrophages. [41, 44] Macrophages were prepared from isolated monocytes by incubation in the presence of M-CSF (100 ng/ml) for 7 days in Teflon beakers, as previously described.[45]
Quantitative Real-Time rt-PCR
Total cell RNA was isolated from 5 x 106 monocytes or macrophages and 12 x 106 neutrophils using the RNeasy Minikit 50 according to instructions by the manufacturer (Qiagen, Valencia CA). DNAfree (Ambion Inc, Austin, TX) was used to remove contaminating DNA. Reverse transcription to prepare cDNA was performed with random primers (Amersham Pharmacia Biotech, Piscataway, NJ) and Superscript II (Life Technologies, Carlsbad, CA). RNAseOUT (Life Technologies, Carlsbad, CA) was used to inhibit ribonuclease activity during reaction sequences. mRNA and cDNA samples were quantified by UV spectroscopy measurements. The PCR reaction volume was 20 μl containing 6 μl cDNA sample, 10 μl 2X Taqman Universal PCR Master Mix (PE Applied Biosystems, Foster City, CA), 0.1 μl of each primer (Biosearch Technologies, Inc., Novato, CA), 0.225 μl fluorogenic probe (Biosearch Technologies), and 3.575 μl DNAase free water. The oligonucletoide primers for respective TLRs were CTGCAAGCTGCGGAAGATAAT, TLR2; AGAGTTTCCTGCAATGGATCAAG, TLR4; GGCTTAATCACACCAATGTCACTATAG, TLR5; and TCTGAAGACTTCAGGCCCAACT, TLR9 for forward primers, and GCAGCTCTCAGATTTACCCAAAA, TLR2; TTATCTGAAGGTGTTGCACATTCC, TLR4; TTAAGACTTCCTCTTCATCACAACCTT, TLR5; and TGCACGGTCACCAGGTTGT, TLR9 for the reverse primers. The fluorogenic probes were CCGCTGAGCCTCGTCCATGGG, TLR2; TTCGTTCAACTTCCACCAAGAGCTGCCT, TLR4; TACACACAATATATGTCTGCAGGAGGCCCA, TLR5; and AGCACCCTCAACTTCACCTTGGATCTGTC, TLR 9. A GeneAmp 5700 Sequence Detection System (PE Applied Biosystems) was used to complete the PCR reactions and monitor chain elongation. Relative expression of TLRs was normalized to expression of 18s RNA. The final normalized results were calculated by dividing the relative transcript levels of the test genes by the relative amount of the 18s RNA.
Statistical analysis
Expression levels are expressed as means +/- SD. Comparisons between multiple groups were performed by factorial analysis of variance (ANOVA). A p value < 0.05 was considered significant.
Results
Relative constitutive expression of TLR2, TLR4, TLR5, and TLR9 in normal human neutrophils, monocytes, and monocyte-derived macrophages
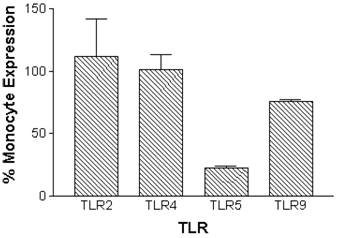
Constitutive mRNA expression was measured in monocytes and neutrophils immediately after cell isolation procedures. Expression of TLR2, TLR4, TLR5 and TLR9 was detectable in normal human peripheral blood monocytes. Neutrophils had similar relative constitutive expression of TLR2, TLR4 and TLR9, but less TLR5, compared to monocytes (Figure 1).
Relative constitutive expression of neutrophil TLR2, TLR4, TLR5, and TLR9 expressed as percentages of monocyte expression of these TLRs in normal human monocytes and neutrophils. mRNA was prepared from 5x106 monocytes (n=10 independent healthy volunteers) or 12x106 neutrophils (n=5 independent healthy volunteers) immediately following isolation of cells. Quantitative real time rt-PCR technology was used to determine relative expression of TLRs normalized to the expression of 18s.
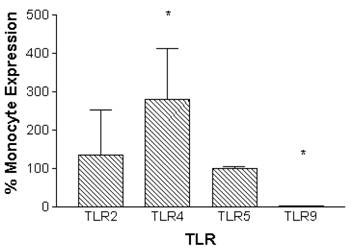
Monocytes can be differentiated into macrophages in vitro through incubation in the presence of M-CSF for 7 days.[46] Macrophages that were derived in vitro constitutively expressed TLR2 and TLR5 at similar levels compared to monocytes. In contrast, TLR4 expression was significantly increased by 280 ± 134 %, and TLR9 expression was decreased to barely detectable levels when compared to monocytes (Figure 2).
Relative constitutive expression of monocyte-derived macrophage TLR2, TLR4, TLR5, and TLR9 expressed as percentages of monocyte expression of these TLRs. mRNA was prepared from 5x106 monocytes (n=10 independent healthy volunteers) immediately following isolation of cells. Macrophages were prepared from monocytes by incubation in the presence of M-CSF (100ng/ml) for 7 days. mRNA was prepared from 5x106 macrophages (n=6 independent healthy volunteers). Quantitative real time rt-PCR technology was used to determine relative expression of TLRs normalized to the expression of 18s. Repeated measures ANOVA was used for statistical analysis. * indicate statistical significance with P<0.05.
Modulation of TLR2, TLR4, TLR5, and TLR9 expression in normal human monocytes by IFN-γ, GM-CSF, and M-CSF
Monocytes were isolated and purified from human peripheral blood and then incubated in the presence of stimulatory cytokines. Incubation with IFN-γ for 3 hours up-regulated expression of TLR2 and TLR4 and down-regulated TLR5 expression in monocytes. TLR9 expression was not affected by IFN-γ (Figure 3).
Modulation of TLR expression in normal human monocytes by IFN-γ, GM-CSF, and M-CSF following incubation for 3-hours. mRNA was prepared from 5x106 monocytes immediately after cell isolation (constitutive expression) or after a three hour incubation in the presence of IFN-γ (103 units/ml), GM-CSF (100ng/ml), or M-CSF (100ng/ml). n=4 healthy normal human donors for TLR2 and TLR4. n=3 normal healthy donors for TLR5 and TLR9. Quantitative real time rt-PCR technology was used to determine relative expression of TLRs normalized to the expression of 18s. Repeated measures ANOVA was used for statistical analysis. * indicate statistical significance with P<0.05.
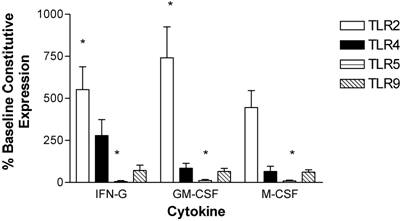
Following a 3-hour incubation with GM-CSF, monocytes had increased expression of TLR2 and decreased expression of TLR5. TLR2 was significantly increased by 740 ± 180 %. TLR4 and TLR9 levels were not affected by GM-CSF (Figure 3).
Monocyte TLR2 and TLR5 expression was also affected by a 3-hour incubation with M-CSF. TLR2 expression was up-regulated by 450 ± 100 %. TLR5 was down-regulated, while expression of TLR4 and TLR9 was not altered (Figure 3).
After a 24-hour incubation, LPS had a robust effect on the expression of monocyte TLR2, with upregulation by 450 ± 160%. IFN-γ and GM-CSF did not maintain increased expression of TLR2 as seen after the 3-hour incubation, and the levels of expression had returned to the baseline constitutive expression of unstimulated monocytes at time zero. M-CSF caused a trend toward increased TLR2 expression and maintained TLR5 levels at initial constitutive levels. IFN-γ, GM-CSF and LPS all led to decreased levels of TLR5. TLR9 expression was at initial constitutive levels at 24 hours following incubation with each of the cytokines whereas LPS stimulation resulted in a trend to reduced levels (Figure 4).
Modulation of TLR expression in normal human monocytes by IFN-γ, GM-CSF, M-CSF or LPS following incubation for 24-hours expressed as percentages of constitutive monocyte expression at time zero. mRNA was prepared from 5x106 monocytes immediately after cell isolation (constitutive expression) or following a 24-hour incubation in the absence or presence of IFN-γ (103 units/ml), GM-CSF (100ng/ml), M-CSF (100ng/ml) or LPS (1μg/ml). n=4 healthy normal human donors for TLR2 and TLR9. n=5 normal healthy donors for TLR4 and TLR5. Quantitative real time rt-PCR technology was used to determine relative expression of TLRs normalized to the expression of 18s. Repeated measures ANOVA was used for statistical analysis. * indicate statistical significance with P<0.05.
Modulation of TLR2, TLR4, TLR5, and TLR9 expression in normal human neutrophils by IFN-γ, GM-CSF, and G-CSF
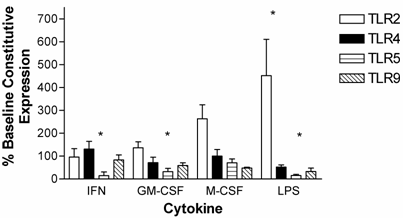
Human peripheral blood neutrophils were evaluated for changes in expression of TLR2, TLR4, TLR5, and TLR9 following incubation with pro-inflammatory cytokines. Short term (3-hour) incubation with IFN-γ up-regulated expression of TLR2, TLR4 and TLR5 in neutrophils by 237 ± 80 %, 307 ± 87 % and 417 ± 130 %, respectively. In contrast, IFN-γ down-regulated TLR9 expression in neutrophils (Figure 5).
Modulation of TLR expression in normal human neutrophils by IFN-γ, G-CSF, and GM-CSF following incubation for 3-hours expressed as percentages of constitutive neutrophil expression. mRNA was prepared from 12x106 neutrophils immediately after cell isolation (constitutive expression) or after a three hour incubation in the presence of IFN-γ (103 units/ml), G-CSF (100ng/ml), or GM-CSF (100ng/ml). n=4 healthy normal human donors for TLR2 and TLR4. n=3 normal healthy donors for TLR5 and TLR9. Quantitative real time rt-PCR technology was used to determine relative expression of TLRs normalized to the expression of 18s. Repeated measures ANOVA was used for statistical analysis. * indicate statistical significance with P<0.05.
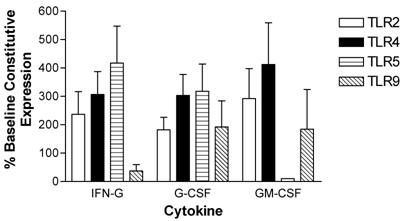
A 3-hour incubation with G-CSF up-regulated expression of all four TLRs in neutrophils (Figure 5). Specifically, G-CSF induced up-regulation of TLR2 expression by 182 ± 44 %, TLR 4 expression by 303 ± 74 %, TLR5 expression by 318 ± 96 %, and TLR9 expression by 192 ± 92 %.
GM-CSF also strongly up-regulated neutrophil TLR2 (292 ± 105 %) and TLR4 412 ± 147 %) expression in neutrophils. TLR9 expression was up-regulated to a lesser extent. In contrast to IFN-γ and G-CSF, GM-CSF induced a strong down-regulation in TLR5 expression after a 3-hour incubation (Figure 5).
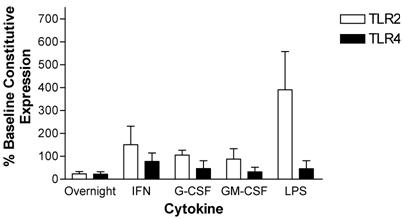
Neutrophil TLR expression patterns were also measured after a 24-hour incubation. Cytokine stimulated cell preparations were compared to both baseline (time zero) constitutive expression and unstimulated cells at 24hours since neutrophils undergo rapid apoptosis soon after isolation. After a 24-hour incubation, expression of TLR2 and TLR4 declined in neutrophils maintained in culture in the absence of cytokine supplementation. Specifically, TLR2 and TLR4 expression decreased by 76 ± 9.5 % and 78 ± 10 %, respectively. Incubation with G-CSF, GM-CSF, or LPS did not affect the decline in TLR4 expression. In contrast, TLR4 expression was maintained at baseline constitutive levels after incubation with IFN-γ. TLR2 expression was maintained at initial constitutive levels when neutrophils were incubated for 24 hours in the presence of IFN-γ, G-CSF or GM-CSF. LPS increased TLR2 expression above the initial constitutive expression level by 390 ± 170 % (Figure 6).
Modulation of TLR expression in normal human neutrophils by IFN-γ, G-CSF, GM-CSF or LPS following incubation for 24-hours expressed as percentages of constitutive neutrophil expression. mRNA was prepared from 12x106 neutrophils immediately after cell isolation or following a 24-hour incubation in the absence or presence of IFN-γ (103 units/ml), G-CSF (100ng/ml), GM-CSF (100ng/ml) or LPS (1μg/ml). n=3 healthy normal human donors for TLR2 and TLR4. Quantitative real time rt-PCR technology was used to determine relative expression of TLRs normalized to the expression of 18s. Repeated measures ANOVA was used for statistical analysis. * indicate statistical significance with P<0.05.
Discussion
Previous studies have shown that human TLRs are differentially expressed in a variety of tissues.[16, 17, 22, 23] This study examined the expression of TLR2, TLR4, TLR5, and TLR9 in normal primary human professional phagocytes. Constitutive expression of TLR2, TLR4, and TLR9 was found to be similar in monocytes and neutrophils, while neutrophils expressed relatively less TLR5 compared to monocytes. Normal human monocyte-derived macrophages had similar expression levels of TLR2 and TLR5 when compared to monocytes. In contrast, relative TLR4 expression was increased in macrophages, and TLR9 expression could not be detected in macrophages. Although previous studies have reported expression of these TLRs in human neutrophils, monocytes, and/or macrophages, the present study is the first to describe and directly compare relative expression levels in all 3 primary cell types.[22, 23, 37, 38]
Various stimuli have been shown to regulate expression of TLRs in leukocytes, including pathogen structures and TLR ligands, such as araLAM and LPS, and pro-inflammatory cytokines, including IL-1β, TNF-α, GM-CSF and IFN-γ.[16, 22, 37, 38] It is plausible that an advantage in pathogen recognition is conferred by controlled expression of germline-encoded pathogen recognition receptors by pro-inflammatory signaling. Recent studies revealed that expression of both TLR2 and TLR4 is increased in monocytes and neutrophils from human patients with sepsis. [47, 48] In these studies, ligands for either TLR2 or TLR4 failed to directly affect expression of TLR2 or TLR4 in monocytes and neutrophils from either septic patients or controls, suggesting the possibility that increased expression of TLRs in this setting may be mediated via by pro-inflammatory cytokines rather than TLR-ligand binding per se.
The results of our study demonstrate that several pro-inflammatory cytokines contribute to the regulation of TLR expression. Of the cytokines studied, IFN-γ and GM-CSF caused the most robust effects on TLR expression with statistically significantly increased TLR2 expression in monocytes, a trend for increased TLR4 in monocytes (IFN-γ) and trends for increased TLR2 and TLR4 in neutrophils. These cytokines have been shown to increase the microbicidal activity of monocytes and neutrophils. [42] We hypothesize that the effects on TLR expression by these cytokines contribute to the enhanced antimicrobial activity. Our study cannot evaluate this directly but previous publications support the premise that increased TLR expression enhances the effector cell response to TLR ligands.
Several studies have identified an association between increased inflammatory activity in monocytes and neutrophils after TLR expression modulation by IFN-γ or GM-CSF. IFN-γ-primed monocytes had increased TLR4 expression and also increased responses to LPS, as measured by NF-κB DNA binding activity and cytokine production (e.g., TNF-α).[39] Neutrophils had increased protein and mRNA expression of TLR2 and TLR9 after incubation with GM-CSF.[38] Furthermore, neutrophils pre-incubated with GM-CSF had increased inflammatory activity, as measured by IL-8 production due to signaling through various TLR ligands.[38] In another study, GM-CSF increased TLR2-mediated cytokine and superoxide anion production in neutrophils.[37] In vivo models have also demonstrated an association between TLR expression levels controlled by proinflammatory cytokines and increased responses to TLR ligands. GM-CSF was shown to play an important role in inflammatory signaling in a model of LPS-induced lung injury. Pretreatment of mice with an inhibitor of GM-CSF prior to LPS instillation caused decreased TLR4 mRNA expression, associated with decreased lung neutrophil and macrophage infiltration and decreased levels of TNF- α and macrophage inflammatory protein-2 (MIP2). [49] In another study using transgenic mice with varyingTLR4 DNA copy number, mice with increased TLR4 DNA copies had increased lung inflammatory responses to intranasally administered LPS. [50]
While TLR2 and TLR4 were up-regulated by GM-CSF and IFN-γ in monocytes, TLR5 expression was diminished by these cytokines in monocytes and by GM-CSF in neutrophils. These results may highlight the relative importance of TLR2 and TLR4 in anti-bacterial innate immunity surveillance. Alternatively, they suggest that separate expression control of TLR5 by inflammatory cytokines may contribute to regulation of innate immunity in monocytes and neutrophils.
The effects of G-CSF and M-CSF on TLR expression in neutrophils and monocytes were less striking. Neutrophil surface TLR2 has been shown to be increased by G-CSF in previous studies.[37] The present study demonstrated a small increase in expression of TLR2 mRNA in neutrophils. Unlike IFN-γ and GM-CSF, only G-CSF led to increased levels of both TLR5 and TLR9 in neutrophils.
This study did not evaluate the effects of immune-modulating cytokines on protein expression but focused on the effects on TLR mRNA expression. Proinflammatory cytokines may also control protein expression of TLRs on the cell surface and intracellularly through regulation of inhibitory RNA pathways, protein translation and post-translational processes. Our results show that TLR mRNA expression is regulated by these cytokines, and we speculate that this may contribute to enhanced cellular responses to pathogens.
The human TLR family is a recently described component of the human innate immune system, and our knowledge of the regulation of these pathogen-discriminating receptors continues to evolve. This report demonstrates that cytokines with known ability to enhance microbicidal activity of phagocytes differentially regulate TLR2, TLR4, TLR5 and TLR9 mRNA expression in neutrophils and monocytes. IFN-γ strongly increased the expression of the bacterial recognition receptors TLR2 and TLR4 on both neutrophils and monocytes. GM-CSF treatment also increased expression of TLR2 and TLR4 on neutrophils and has previously been shown to enhance TLR ligand responses by these cells. These results demonstrate that up-regulated TLR expression may be a component of cytokine-mediated enhancement of phagocyte-mediated host defense and suggest a promising role for IFN-γ and/or GM-CSF as therapeutic immunomodulators for enhancing bacterial recognition by the innate immune system.
Acknowledgements
This study was supported in part by a Magnuson Scholar Fellowship from the University of Washington (DSO), RO1 HL62995 from the National Institutes of Health (WCL), and a Canada Research Chair in Infectious Diseases and Inflammation (WCL).
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Medzhitov R, Janeway C Jr. Innate immunity. N Engl J Med. 2000;343(5):338-44
2. Medzhitov R, Janeway C Jr. The Toll receptor family and microbial recognition. Trends Microbiol. 2000;8(10):452-6
3. Roach JC, Glusman G, Rowen L. et al. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci U S A. 2005;102(27):9577-82
4. Chuang T, Ulevitch RJ. Identification of hTLR10: a novel human Toll-like receptor preferentially expressed in immune cells. Biochim Biophys Acta. 2001;1518(1-2):157-61
5. Chuang TH, Ulevitch RJ. Cloning and characterization of a sub-family of human toll-like receptors: hTLR7, hTLR8 and hTLR9. Eur Cytokine Netw. 2000;11(3):372-8
6. Chaudhary PM, Ferguson C, Nguyen V. et al. Cloning and characterization of two Toll/Interleukin-1 receptor-like genes TIL3 and TIL4: evidence for a multi-gene receptor family in humans. Blood. 1998;91(11):4020-7
7. Du X, Poltorak A, Wei Y, Beutler B. Three novel mammalian toll-like receptors: gene structure, expression, and evolution. Eur Cytokine Netw. 2000;11(3):362-71
8. Takeuchi O, Kawai T, Sanjo H. et al. TLR6: A novel member of an expanding toll-like receptor family. Gene. 1999;231(1-2):59-65
9. Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci U S A. 1998;95(2):588-93
10. Ozinsky A, Underhill DM, Fontenot JD. et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97(25):13766-71
11. Underhill DM, Ozinsky A, Hajjar AM. et al. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401(6755):811-5
12. Mukhopadhyay S, Herre J, Brown GD, Gordon S. The potential for Toll-like receptors to collaborate with other innate immune receptors. Immunology. 2004;112(4):521-30
13. Medzhitov R, Janeway C Jr. Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89-97
14. Belvin MP, Anderson KV. A conserved signaling pathway: the Drosophila toll-dorsal pathway. Annu Rev Cell Dev Biol. 1996;12:393-416
15. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499-511
16. Flo TH, Halaas O, Torp S. et al. Differential expression of Toll-like receptor 2 in human cells. J Leukoc Biol. 2001;69(3):474-81
17. Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168(2):554-61
18. Parker LC, Whyte MK, Dower SK, Sabroe I. The expression and roles of Toll-like receptors in the biology of the human neutrophil. J Leukoc Biol. 2005;77(6):886-92
19. Heil F, Ahmad-Nejad P, Hemmi H. et al. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol. 2003;33(11):2987-97
20. Matsumoto M, Funami K, Tanabe M. et al. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol. 2003;171(6):3154-62
21. Ahmad-Nejad P, Hacker H, Rutz M, Bauer S, Vabulas RM, Wagner H. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur J Immunol. 2002;32(7):1958-68
22. Muzio M, Bosisio D, Polentarutti N. et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol. 2000;164(11):5998-6004
23. Kokkinopoulos I, Jordan WJ, Ritter MA. Toll-like receptor mRNA expression patterns in human dendritic cells and monocytes. Mol Immunol. 2005;42(8):957-68
24. Seya T, Funami K, Taniguchi M, Matsumoto M. Antibodies against human Toll-like receptors (TLRs): TLR distribution and localization in human dendritic cells. J Endotoxin Res. 2005;11(6):369-74
25. Peng G, Guo Z, Kiniwa Y. et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309(5739):1380-4
26. Hajjar AM, O'Mahony DS, Ozinsky A. et al. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J Immunol. 2001;166(1):15-9
27. Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163(1):1-5
28. Takeuchi O, Hoshino K, Kawai T. et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443-51
29. Aliprantis AO, Yang RB, Mark MR. et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285(5428):736-9
30. Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A. 1999;96(25):14459-63
31. Lien E, Sellati TJ, Yoshimura A. et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem. 1999;274(47):33419-25
32. Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem. 1999;274(25):17406-9
33. Poltorak A, He X, Smirnova I. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085-8
34. Hoshino K, Takeuchi O, Kawai T. et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162(7):3749-52
35. Bauer S, Kirschning CJ, Hacker H. et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A. 2001;98(16):9237-42
36. Hayashi F, Smith KD, Ozinsky A. et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410(6832):1099-103
37. Kurt-Jones EA, Mandell L, Whitney C. et al. Role of toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF enhances TLR2 expression and TLR2-mediated interleukin 8 responses in neutrophils. Blood. 2002;100(5):1860-8
38. Hayashi F, Means TK, Luster AD. Toll-like receptors stimulate human neutrophil function. Blood. 2003;102(7):2660-9
39. Bosisio D, Polentarutti N, Sironi M. et al. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood. 2002;99(9):3427-31
40. Mita Y, Dobashi K, Shimizu Y, Nakazawa T, Mori M. Toll-like receptor 2 and 4 surface expressions on human monocytes are modulated by interferon-gamma and macrophage colony-stimulating factor. Immunol Lett. 2001;78(2):97-101
41. Liles WC. Immunomodulatory approaches to augment phagocyte-mediated host defense for treatment of infectious diseases. Semin Respir Infect. 2001;16(1):11-7
42. Hubel K, Dale DC, Liles WC. Therapeutic use of cytokines to modulate phagocyte function for the treatment of infectious diseases: current status of granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, macrophage colony-stimulating factor, and interferon-gamma. J Infect Dis. 2002;185(10):1490-501
43. Klebanoff SJ, Olszowski S, Van Voorhis WC, Ledbetter JA, Waltersdorph AM, Schlechte KG. Effects of gamma-interferon on human neutrophils: protection from deterioration on storage. Blood. 1992;80(1):225-34
44. Gaviria J, van Burik J, Dale D, Root R, Liles W. Comparison of interferon-gamma, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor for priming leukocyte-mediated hyphal damage of opportunistic fungal pathogens. J Infect Dis. 1999;179(4):1038-41
45. Kiener PA, Davis PM, Starling GC. et al. Differential induction of apoptosis by Fas-Fas ligand interactions in human monocytes and macrophages. J Exp Med. 1997;185(8):1511-6
46. Young DA, Lowe LD, Clark SC. Comparison of the effects of IL-3, granulocyte-macrophage colony-stimulating factor, and macrophage colony-stimulating factor in supporting monocyte differentiation in culture. Analysis of macrophage antibody-dependent cellular cytotoxicity. J Immunol. 1990;145(2):607-15
47. Armstrong L, Medford AR, Hunter KJ, Uppington KM, Millar AB. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol. 2004;136(2):312-9
48. Harter L, Mica L, Stocker R, Trentz O, Keel M. Increased expression of toll-like receptor-2 and -4 on leukocytes from patients with sepsis. Shock. 2004;22(5):403-9
49. Bozinovski S, Jones J, Beavitt SJ, Cook AD, Hamilton JA, Anderson GP. Innate immune responses to LPS in mouse lung are suppressed and reversed by neutralization of GM-CSF via repression of TLR-4. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L877-85
50. Togbe D, Schnyder-Candrian S, Schnyder B. et al. TLR4 gene dosage contributes to endotoxin-induced acute respiratory inflammation. J Leukoc Biol. 2006;80(3):451-7
Author contact
![]() Correspondence to: W. Conrad Liles, MD, PhD, Professor and Vice-Chair of Medicine, Director, Division of Infectious Diseases, Toronto General Hospital, 13E 220, 200 Elizabeth Street, Toronto, ON M5G 2C4, CANADA. Ph: 416-340-4800 x3624; FAX: 416-340-3357; Email: conrad.lileson.ca
Correspondence to: W. Conrad Liles, MD, PhD, Professor and Vice-Chair of Medicine, Director, Division of Infectious Diseases, Toronto General Hospital, 13E 220, 200 Elizabeth Street, Toronto, ON M5G 2C4, CANADA. Ph: 416-340-4800 x3624; FAX: 416-340-3357; Email: conrad.lileson.ca