Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2006; 3(1):26-27. doi:10.7150/ijms.3.26 This issue Cite
Case Report
Hb J- Meerut [α 120 (H3) Ala ->Glu (α1)] In A Turkish Male
Gunçag Dinçol1, Serkan Güvenç1, Dedrey Elam2, Abdullah Kutlar2, Ferdane Kutlar2 ![]()
1. Division of Hematology, Department of Internal Medicine, Istanbul Medical School, University of Istanbul, Çapa, Istanbul, Turkey
2. Titus H.J. Huisman Hemoglobinopathy Laboratory, Department of Medicine, Medical College of Georgia, Augusta, Georgia, USA
Received 2005-12-3; Accepted 2006-1-28; Published 2006-2-2
Abstract
Hb J Meerut is an infrequently found α-globin variant. It has previously been reported in various populations around the world. One particular case reported in 1994 included a Turkish family. In this report, details of a second case of Hb J Meerut in a Turkish male who is unrelated to the first family are described. In the present case a slight increase in the oxygen affinity of Hb J Meerut, relative to that of the normal control, has been observed as detected by low p50 values in arterial whole blood. Additionally, a slight increase in red blood cell count, as compared against a normal individual, was observed.
Keywords: Hb J-Meerut [α 120 (H3) Ala->Glu (α1)], DNA analysis, Turkish male, slightly increased oxygen affinity
1. INTRODUCTION
Hb J-Meerut results from a C ->A mutation (GCG->GAG) at codon 120 of the α1 or α2 globin gene, changing the alanine to glutamic acid at residue 120 of the α chain [1,2,3]. This variant was first reported in two sisters from Meerut, Utlar Pradesh, India [1] and in two brothers from Bangladesh living in Birmingham, England [2]; subsequently the same abnormal hemoglobin, was described in one Japanese family [4] and in one Turkish family [5]. The present study provides details about α Hb J Meerut heterozygous Turkish male who is unrelated to the family with the same abnormal hemoglobin described previously from Turkey.
2. A CASE REPORT AND RESULTS
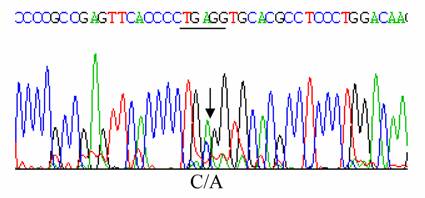
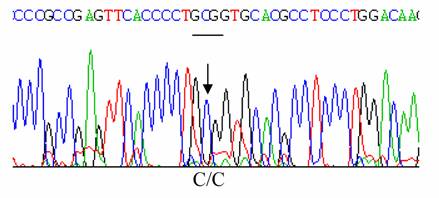
The propositus was a healthy 34-year old male native of Isparta, a city situated in Western Turkey. Informed consent was obtained from the patient. He had no symptoms attributable to a hemolytic process. Hematological data were as follows: Hb 16.9 g/dl, RBC 5.8x1012/L; PCV 0.49 L/L, MCV 84 fL, MCH 29 pg; MCHC 34.4 g/dL, reticulocytes 1 %. An abnormal hemoglobin with a mobility similar to that of Hb J was detected by cellulose acetate electrophoresis at pH 8.6, and had same electrophoretic mobility with Hb A by citrate agar electrophoresis at pH 6.2 [6]. Modified chromatographic analysis of red cell lysate was done by HPLC using a cation exchange column. The column was an Ion-Exchange Cartridge column, 0.59x3.6 cm manufactured by BioRad and obtained from MedTex Company, Istanbul, Turkey. The chromatogram was developed with sodium phosphate and sodium azide buffers. Abnormal Hb was 20.0 % of the total Hb; HbA2; 2.0% and Hb A ;77.3% [6]. HbF value was determined by alkali denaturation method and found as 0.7 % [7]. The p50 values obtained (using the Radiometer ABL 700; Radiometer ABL, Copenhagen, Denmark) at pH 7.4 and at 37 °C, were 24.96 mmHg for a whole arterial blood sample from propositus and 28.67 mmHg for that of the normal control [6]. The result of an Isopropanol stability test was negative [6,8]. Ten ml of peripheral blood, collected with EDTA as the anticoagulant, were sent for structural DNA analysis by overnight express courier to the Titus H.J. Huisman Hemoglobinopathy Laboratory, Medical College of Georgia, Augusta, GA, USA. DNA was extracted from peripheral blood leukocytes as previously described by Poncz et al [9]. The α1 - and α2- globin genes were separately amplified as described before [10]. Polymerase chain reaction (PCR) products were then purified with the Prep-A Gene DNA purification Kit (Bio-Rad Laboratories, Hercules, CA, USA) and subjected to cycle sequencing with the BDT (Big Dye Terminator) method on an ABI PRISMTM 377 Cycle Sequencer, according to manufacturer's instructions (Applied BioSystems Inc., Foster City, CA, USA) at the Molecular Biology Core Facility, Medical College of Georgia, Augusta, GA, USA. Sequencing of the α2-globin gene did not reveal any abnormality, shown in Figure 2. However, nucleotide sequencing of the α1-globin gene showed a C ->A mutation at codon 120 in exon–3, thus identifying the variant as Hb J-Meerut [ α120 (H3) Ala -> Glu], as illustrated in Figure 1.
Sequencing of α1 globin gene with a mutation at codon 120 (GCG->GAG)
Sequencing of α2 globin gene with no mutation at codon 120 (GCG)
Complete nucleotide sequence of the α1 globin gene was submitted to the GenBank (access # AY196787).
3. DISCUSSION
A GCG ->GAG mutation was found in codon 120 of both α1 and α2 globin genes [3]. In general, the average percentage of the abnormal hemoglobin in heterozygote with α1 mutations (19.7 %) was slightly lower than that in heterozygote with α2 mutations (23.5 %). This decrease applies to stable hemoglobins only [3]. Position α120 is external and is not involved in heme binding or subunit contacts but is involved in the α1β1 contacts in Hb molecule [11,12]. The amino acid substitution at this site may be expected to cause no abnormalities for oxygenation; however, the measurement of the oxygen equilibrium curves of Hb J Meerut showed a slightly increased oxygen affinity [4]. In our case, the slight increase in the oxygen affinity of Hb J Meerut relative to that of the normal control has been shown by the low p50 values in arterial whole blood. In Hb J Meerut of glutamic acid residue replaced by alanine residue at α120 might interact with the side chain of arginine residue at β 30 of one of the two β chains to form a weak salt bridge, thereby causing a slightly increased oxygen affinity.
Conflict of interests
The authors have declared that no conflict of interest exists.
References
1. Blackwell RQ, Boon WH, Wang CL, Weng MI, Liu CS. Hemoglobin J Meerut: α120 Ala ->Glu. Biochim Biophys Acta. 1974 ;351:7-12
2. Kamuzora H, Lehmann H, Griffiths KD, Mann JR, Raine DN. A new hemoglobin variant hemoglobin J Birmingham α120 (H3) Ala ->Glu. Ann clin biochem. 1974 ;11:53-55
3. Molchanova TP, Pobedimskaya DD, Huisman THJ. The differencs in quantities of α2- and α1-globin gene variants in heterozygotes. Br J Haematol. 1994 ;88:300-306
4. Harano T, Harano K, Imai K, Yunoki H, Yagi H, Nagashima K, Kuroume T. Hb J-Meerut [α120 (H3) Ala ->Glu] found in a Japanese family. Hemoglobin. 1989 ;13(2):169-175
5. Yalçin A, Avci F, Beyhan C, Gürgey A, Ural AU. A case of Hb J-Meerut (or Hb J-Birmingham) [α 120 (H3) Ala ->Glu]. Hemoglobin. 1994 ;18(6):433-435
6. Kutlar A, Huisman THJ. Detection of hemoglobinopathies. In: (ed.) Hommes FA. Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual. New York: Wiley Liss Inc. 1991:519-560
7. Singer K, Chernoff Al, Singer L. Studies on abnormal hemoglobins: their demonstration in sickle cell anemia and other hematologic disorders by means of alkali denaturation. Blood. 1951 ;6:413-428
8. Carrell RW, Kay R. A simple method for the detection of unstable hemoglobins. Br J Haematol. 1972 ;23(5):615-619
9. Poncz M, Solowiejczyk D, Harpel B, Mory Y, Schwartz E, Surrey S. Construction of human gene libraries form small amounts of peripheral blood: analysis of β -like globin genes. Hemoglobin. 1982 ;6(1):27-46
10. Molchanova TP, Pobedimskaya DD, Postnikov Y. A simplified procedure for sequencing amplified DNA containing the α2- or α1- globin gene. Hemoglobin. 1994 ;18(3):251-255
11. Sack JS, Andrews LC, Magnus KA, Hanson JC, Rubin J, Love WE. Location of amino acid residues in human deoxy hemoglobin. Hemoglobin. 1978 ;2(2):153-169
12. Perutz MF, Lehmann H. Molecular Pathology of Human Hemoglobin. Nature. 1968 ;219:902-909
Author contact
![]() Corresponding address:
Corresponding address:
Ferdane Kutlar MD, Titus H.J. Huisman Hemoglobinopathy Laboratory, Department of Medicine, Medical College of Georgia, Augusta, Georgia, USA. FKUTLARmcg.edu