Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. From Acute to Chronic HBV...
2. Relevance and Utility of HBV...
3. HBV Treatment Options and...
4. Molecular Mechanisms of Drug...
5. Monitoring HBV Drug Resistance
6. Management of HBV Drug...
7. Clinical Impact of Hepatitis...
8. Research Directions
References
Tables and Figures
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2005; 2(1):8-16. doi:10.7150/ijms.2.8 This issue Cite
Review
Advances in Molecular Diagnosis of HBV Infection and Drug Resistance
Erwin Sablon, Fred Shapiro ![]()
Innogenetics NV, Industriepark Zwijnaarde, Belgium
Received 2004-10-1; Accepted 2005-1-2; Published 2005-1-5
Abstract
Serological markers are key elements in diagnosing acute hepatitis B virus (HBV) infection and determining its possible evolution towards chronicity. Once treatment of chronic HBV is initiated with approved anti-hepadnaviral agents, such as lamivudine, interferon-alpha, or adefovir dipivoxil, the measurement of HBV DNA in serum can not only help monitor treatment efficacy but also indicates breakthrough infection should drug resistance emerge. Advances in the molecular diagnosis of drug resistance using highly sensitive methodologies such as DNA hybridization assays can further pinpoint the type of mutation responsible and, more importantly, detect upcoming viral resistance at an early stage when the variant represents only a minor fraction of the total viral population. Such new tools are especially relevant for patients at high risk for disease progression or acute exacerbation. Recent diagnostic developments including HBV genotyping and precore/core promoter assays that could well play important future roles in HBV patient management are also reviewed.
Keywords: Molecular Diagnosis, HBV Infection, Drug Resistance
1. From Acute to Chronic HBV Disease
When hepatitis B virus (HBV) infection is suspected, it is the appearance of serum markers of HBV infection that establishes the diagnosis of the disease. Of these, HBsAg is considered to be the sentinel marker for the confirmation of acute infection. Its presence can be detected as early as 6 weeks after exposure, and should therefore be assessed when prodromal (malaise, anorexia, fever, rash, arthralgia) or more classical symptoms (dark urine, jaundice) are observed. Generally, some 8 to 16 weeks will usually have elapsed from exposure to the virus until the appearance of telltale symptoms. HBsAg is also the key marker in determining whether hepatitis B infection has become chronic: defined as the persistence of the surface antigen for at least 6 months [1]. By contrast, if acute disease resolves, HBsAg declines followed by a subsequent rise of antibodies to the surface antigen (anti-HBs) upon recovery (Figure 1).
Evolution of HBV markers in acute and chronic infection.
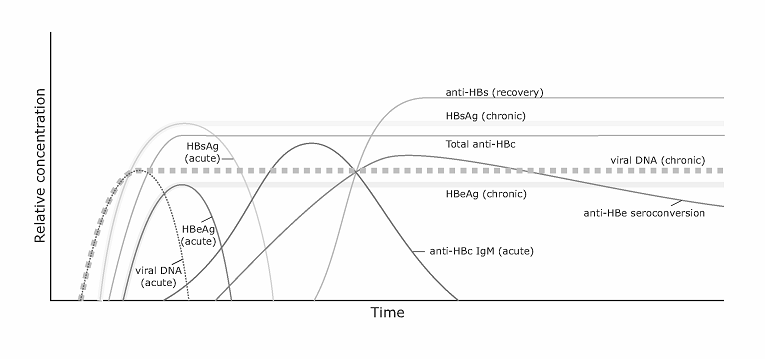
Serum HBV DNA and HBeAg are two other key markers appearing precociously, and are indicative of active viral replication. The former may be present at levels exceeding 105 to 106 copies/mL and can be identified some 6-12 weeks after exposure to the virus, or even earlier if PCR-based methods are used. During acute infection, HBeAg and transaminase levels are also elevated. As the immune system begins to mount its response to infection, an initial rise - then decline - of anti-HBc IgM, is observed. By contrast, anti-HBc IgG rises but persists even after acute infection has resolved. As HBV viremia is cleared, anti-HBe also begins to increase, and alanine aminotransferase (ALT) levels decline [2].
Of the two billion persons worldwide who come into contact with HBV, approximately 6% of the global population (more than 350 million persons) fail to resolve acute viral infection and become chronic carriers, eventually placing between 15% to 25% of such individuals at risk for end-stage liver disease. Should chronic carriage ensue, those persons who are unable to resolve HBV infection enter into a low replication phase of infection marked by the seroconversion of HBeAg to anti-HBe. This change occurs in about 10% (5%-20%) of chronic adult carriers per year [3]. The inactive carrier state is marked by continued HBsAg positivity, in contrast to a drop in HBV DNA levels to less than 105 copies/mL. In addition, ALT levels and anti-HBc IgM decline and normalize; liver histology shows a significant reduction in necro-inflammation. Sometimes the passage to this phase is marked by a flare-up in disease activity termed seroconversion illness [2]. Repeated episodes of active hepatitis may occur until final anti-HBe seroconversion materializes.
This classical course of events may be confounded by several exceptions. For instance, core promoter or precore mutations lead to HBV variants that either drastically curtail or abolish HBeAg production, respectively. As seroconversion to anti-HBe progresses (ie HBeAg-expressing variants are eliminated immunologically, while precore variants escape detection, survive, and increase), circulating levels of HBV DNA remain high and liver disease persists or relapses [4].
Moreover, the age when infection is acquired markedly influences the course of disease. Three predominant patterns have been noted: (i) early (perinatal) infection – especially prevalent in Asia and Oceania – characterized by a long immune tolerant phase (high serum HBV DNA, persistent HBeAg positivity, normal ALT, poor response to therapy); (ii) person-to-person HBV transmission in childhood frequently observed in sub-Saharan Africa, the Mediterranean countries, and Alaska where such children exhibit HBeAg-positivity, elevated ALT, and peripubertal seroconversion to anti-HBe; and (iii) acquisition of HBV during adulthood as often happens among persons in Western countries with an absence of immune tolerance, a relatively rapid progression to immune clearance, and a better response to therapy [1].
2. Relevance and Utility of HBV DNA Detection Methods
Although HBV DNA assays are not presently recommended for the routine evaluation and management of patients with chronic HBV infections, they nevertheless provide very useful adjunct information concerning viral replication – especially in situations when patient serological profiles fall outside of classical patterns. Some key advantages and disadvantages of HBV DNA testing are presented in Table 1.
Advantages and disadvantages of HBV DNA testing
| Advantages | Disadvantages |
| Earliest indicator of infectivity | Definitive role in patient management still to be clarified |
| Can help monitor effectiveness of antiviral therapy | Not yet recommended for routine evaluation |
| Can help assess ongoing disease activity in chronic infections | Not well standardized |
| Can indicate and confirm emergence of antiviral resistance | Wide variation in test sensitivities |
| Direct marker and gold standard for HBV viral replication | No gold standard among different methodologies |
| Useful marker of infectivity in presence of precore/core promoter mutants | Relatively slower to detect drug resistance |
| Confirmation of spontaneous remission or co-infection | Detection of low viral levels of uncertain clinical significance |
| Cut-off levels for inactive disease unclear | |
| Threshold levels for progressive liver disease unknown |
Current HBV DNA assays make use of differing technologies and can generally be divided into (i) signal amplification assays (liquid phase hybridization, antibody capture approach, branched DNA) and (ii) DNA amplification tests based on the polymerase chain reaction (PCR) [for detailed reviews, see 2,5]. Signal amplification assays have sensitivities approaching 1 pg of DNA (105-106 genome copies) or even to 103 genome copies [2]. Alternatively, HBV DNA detection based on a nested PCR approach can detect as few as 102-103 genome copies. At such low titers, problems with contamination and reproducibility may lead to false-positive results, thereby necessitating the use of internal or external standards [5]. Commercial assays that make use of semi-automated systems can overcome these limitations [2,5].
In contrast to PCR tests that measure HBV DNA titers only after completion of the PCR cycle ('endpoint measurement'), real-time PCR technology, based on continuous quantitative monitoring during the exponential phase of the PCR reaction, is able to measure viral loads over a larger dynamic range [2,5]. New developments (TaqMan technology, molecular beacons) that decrease the number of handling steps, reduce contamination, and increase throughput and the accuracy of quantification will further enhance the utility of these assays [2,5].
Given the varied technologies used to determine HBV DNA titers, it is understandable that considerable variation in results may occur when using different viral load tests. This makes standardization of HBV DNA viral load assays an important issue that is yet to be resolved [2]. Collaborative studies have been undertaken to establish international standards for HBV DNA nucleic acid amplification techniques [5,6], but no clear consensus has yet emerged. Finally, the role of viral load testing to detect drug resistance is discussed below.
3. HBV Treatment Options and Drug Resistance
For the 350 million persons chronically infected with HBV, the two therapeutic approaches presently available to control infection and its sequelae are immunomodulatory agents and/or antiviral chemotherapy. Such treatments aim at interrupting the progression and clinical outcomes of the disease (cirrhosis, hepatocellular carcinoma) by stimulating the anti-HBV-specific host immune response or by markedly decreasing viral replication.
The first therapeutic agent to be approved for hepatitis B was interferon-alpha (IFN-α), whose dual mode of action includes both antiviral and immunomodulatory effects. Unfortunately, extended IFN-α treatment is expensive, injection-dependent, effective in no more than 15-25% of patients, and associated with a wide spectrum of adverse reactions [7]. These limitations will be partially obviated by the likely approval of peginterferon-alpha for use in chronic HBV. One recent phase 2, dose-ranging study indicated that the once-weekly dose of this agent resulted in a two-fold higher response rate (HBeAg seroconversion; HBV suppression, ALT normalization) after six months of treatment compared to conventional interferon, although the frequencies of adverse events were similar [8].
However, it is the nucleoside analogue lamivudine that has become the gold standard worldwide for use in patients with chronic hepatitis B. The convenience of its relatively affordable cost, a one-pill-per-day regimen, and the low incidence of side effects has made it the preferred treatment for many patients. Nevertheless, despite a good safety profile and initial efficacy, lamivudine-induced decreases in viral load are difficult to sustain over time due to the occurrence of viral drug resistance. Thus, the antiviral effects of the drug are gradually reversed in most cases. The ensuing rebound effect [9,10] is termed breakthrough infection. It is now known that genotypic resistance to lamivudine emerges in approximately one quarter of patients after one year of treatment [11-13], rising to more than 40% after two years, and increasing further to over 50% and 70% after years three and four, respectively [13]. Increasing the drug dosage does not appear to substantially affect the rate at which viral resistance occurs [11].
The armamentarium of HBV therapy has recently been expanded by the approval of the nucleotide analog, adefovir dipivoxil. Its antiviral efficacy was confirmed in large-scale clinical trials for the therapy of both HBeAg-positive [14] and HBeAg-negative [15] chronic hepatitis B, achieving more than a 3-log decrease in viral load, a significant drop in serum ALT levels, and an improvement in liver histology after one to two years of therapy. Although resistance surveillance in adefovir-treated patients for potential resistance mutations until 48 weeks [16] and then up to 60 weeks [17] did not reveal the emergence of resistant mutants, this did not turn out to be the case upon treatment after 96 weeks [18]. Just as is the present case for lamivudine, resistance testing for adefovir mutations could become necessary should the rate of emergence of resistance and the impact of such mutations turn out to be clinically relevant for therapy management.
4. Molecular Mechanisms of Drug Resistance
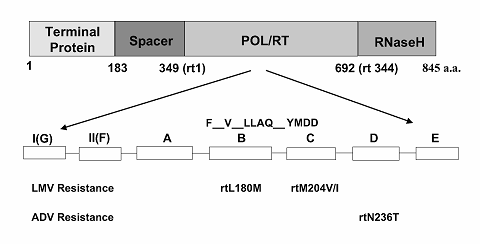
HBV is a small but elusive DNA virus that presents relatively few specific targets for antiviral interventions. At present, the target of choice is the HBV polymerase protein – an enzyme that plays an essential role in viral replication [19]. Within its four functional regions (Figure 2), drug resistance to lamivudine is associated with mutations in the very conserved catalytic polymerase /reverse transcriptase domain of the gene [20], located specifically at a locus of four amino acids consisting of tyrosine-methionine-aspartate-aspartate, termed the YMDD motif [21]. It is thought that lamivudine acts here by suppressing HBV replication. Given that HBV generates up to 1012 virions/day [22], and because of the selective pressure exerted by long-term administration of lamivudine on the virus, HBV mutants emerge.
Key HBV polymerase mutants resistant to lamivudine (LMV) and adefovir dipivoxil (ADV). The HBV polymerase can be divided into four functional regions. Of these, the reverse transcription region (RT), responsible for RNA- and DNA-dependent DNA synthesis contains seven motifs (A thru G).
When mutations occur, the configuration of the wild-type YMDD motif becomes altered in such a way that the drug no longer successfully exerts its inhibitory action at that site. Both wild-type and resistance virus strains then populate the infected liver. HBV DNA and ALT levels usually begin to rebound, but are generally lower compared to baseline levels when only wild-type virus is present.
Three key mutations in the polymerase gene have been shown to confer resistance to lamivudine and adefovir dipivoxil (Figure 2), although many other mutations have also been described [19]. With respect to the recently adopted nomenclature for HBV mutations in the polymerase region [23], the first two include the substitution of methionine (M) by the amino acids isoleucine (I) or valine (V) in the YMDD motif (C domain) at position rtM204V/I. In the majority of cases, these mutations in the YMDD motif occur together with an additional compensatory mutation in the B subdomain [20], namely the substitution of a leucine by methionine some 20 amino acids upstream from the YMDD domain at position rtL180M. Finally, the newly discovered mutant to adefovir (rtN236T) is located downstream from the YMDD motif in the D domain of the viral polymerase.
Assuming correct patient compliance with treatment, resistance to antiviral therapy is presently defined as (i) an increase in serum HBV DNA titers during therapy after a sustained viral response and (ii) the selection of a mutation in the viral polymerase gene (YMDD motif of the polymerase C domain) that could not be detected in the major viral species prior to therapy, and that is not included in the HBV consensus sequences from data banks (i.e., genotypic resistance) [24].
5. Monitoring HBV Drug Resistance
Several options are currently available to monitor HBV drug resistance, and can generally be divided into genotypic assays and phenotypic methods. The former detect changes in the HBV genomic sequence in the course of treatment. In addition, indirect measures of lamivudine resistance including determination of ALT values, and especially HBV DNA (viral load) assays are routinely used in the clinic. A comparison of these methods is presented in Table 2.
Comparison of different commercial and non-commercial testing methods for detection of drug resistance.
| Sensitivity1/Early detection | Information content2 | Updateability3 | Commercial availability | Cost | Complexity of interpretation | Labor intensiveness | Automatability | |
| Genotypic Methods | ||||||||
| Direct Sequencing | 15-50% | high | high | yes | high | high | Intermediate high | yes |
| RFLP | 5-10% | low | low | no | low | intermediate | high | no |
| RT-PCR | 5-10% | low | low | no | high | intermediate | low | no |
| LiPA | 5% | low | low-intermediate | yes | intermediate | low | low | yes |
| Florescence | nd | intermediate | intermediate | no | intermediate | intermediate | intermediate | yes |
| MALDI-TOF | <5% | intermediate | intermediate | No | intermediate | intermediate | intermediate | yes |
| DNA arrays | na | intermediate | intermediate | no | high | high | low | yes |
| Indirect methods | ||||||||
| Viral load | na | na | na | yes | intermediate | low | low | yes |
| Phenotypic Assays | na | na | low | no | high | high | high | no |
1Sensitivity here refers to the lowest level (%) at which an assay can detect mixtures of mutant and wild-type virus. 2Information content is considered as a measure of a test's ability to provide broad, relevant information about possible new mutations. 3Updateability assesses the ease at which a test can be adapted to incorporate the detection of a new mutation. na, not applicable. nd, not determined.
(1) Genotyping Methods
Direct DNA Sequencing
Standard DNA sequencing technology provides highly accurate and complete DNA sequence information, and is applicable to any part of the 3.2-kilobase HBV genome [25]. A serious handicap is the inability of sequencing to detect viral resistance even when the mutated virus still makes up a relatively large fraction (up to 30%) of the entire HBV population (i.e., mixtures of wild-type and mutant species). This limits its use for detecting upcoming resistance at an early stage [26]. Furthermore, it tends to be time-consuming and labor intensive, not readily adaptable to high-throughput screening, and is amenable to analysis only by well-trained personnel.
Clonal Analysis
An additional difficulty when using direct DNA sequencing of a PCR product is to know whether a given set of mutations occurs on the same molecule or in a different clonal subpopulation. This obstacle can theoretically be circumvented by sequencing multiple clones from a given sample. But unless a sufficient amount of clones are analyzed, minor subpopulations may remain undetected [20,27]. The technique is quite laborious, and is not adaptable for large-scale use.
RFLP
This methodology provides a means of overcoming some of the aforementioned limitations. RFLP methodology is as accurate as direct DNA sequencing [28] but, unlike sequencing, can detect samples with mixed virus populations containing mutant virus making up 5% to 10% of the virus population [29,30]. Nevertheless, the procedure is generally long and tedious (multiple PCRs, multiple enzyme digestions), and requires skilled personnel since a specific endonuclease reaction has to be developed for each separate mutation to be analyzed. Such tests tend to be 'home brews,' and have not been commercialized for general use [28,31].
DNA Hybridization
Various types of assays exist that make use of probe-product hybrids. For instance, fluorometric real-time PCR with the LightCycler Assay can be used to detect resistant variants. Samples differing by only one nucleotide can be readily distinguished [32,33]. However, natural sequence variability in places where the fluorescence probes bind can lead to a lower melting temperature of the probe without necessarily being associated with a resistance mutation. Moreover, for unequivocal detection of lamivudine resistance, the variant subpopulation may have to comprise at least 5% to 10% of the total [33]. Another test that employs mixed hybridization-sequencing-PCR (“minisequencing”) technology involves the extension of multiple oligonucleotide primers with fluorescent ddNTPs by means of a DNA polymerase [34].
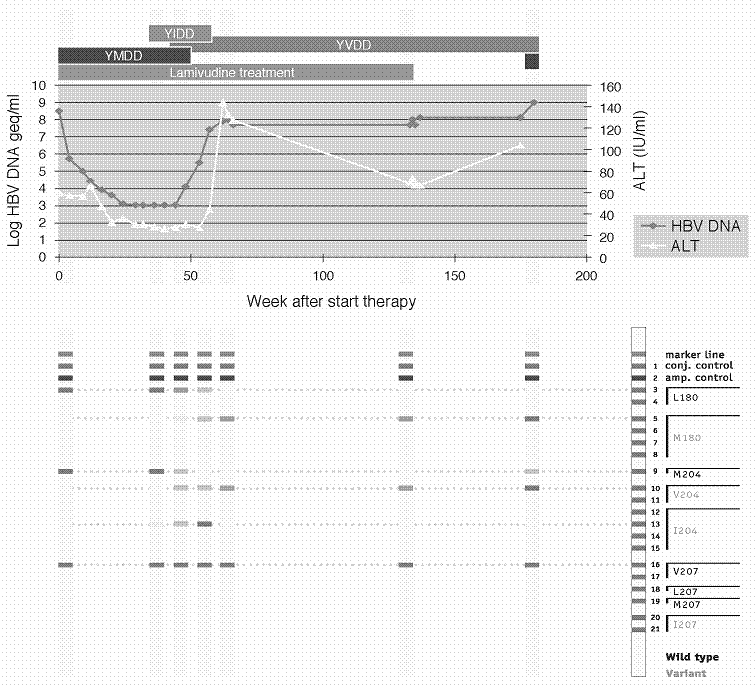
An alternative approach involves differential hybridization to a preexisting panel of membrane-bound probes as illustrated by the commercially available INNO-LiPA HBV DR line probe assay (Figure 3). The test makes use of a series of short immobilized oligonucleotide probes to discriminate between different nucleic acid fragments (down to single nucleotide mismatches), thereby enabling identification of wild-type or mutant variants [23,26]. Importantly, the test can detect variations early during the emergence of viral resistance even when the variant represents only a minor fraction of the total viral population [26,35]. This is especially relevant for patients at high risk for disease progression. An example of the monitoring and early detection of HBV drug resistance with this test is shown in Figure 4. In the consecutive patient samples presented, the YIDD mutation (rtM204I) was detected about 40 weeks after the start of lamivudine therapy, some 10 and twelve weeks before the increases in viral load and ALT, respectively. Subsequently, the YIDD mutation was replaced by a YVDD mutant. This latter mutation was seen to persist (over 40 weeks) after lamivudine treatment was halted.
Example of line probe assay (LiPA) detection system based on the reverse hybridization principle. Such line probe assays make use of a series of short, immobilized oligonucleotides attached as parallel lines on nitrocellulose membrane strips, permitting easy identification of wild-type virus or mutant variants. For this purpose, biotinylated, amplified viral DNA fragments derived from hepatitis B patients are hybridized to the selected immobilized probes. Streptavidin labeled with alkaline phosphatase is then added, and binds to the previously formed biotinylated hybrids. Incubation with a chromogen results in color development. After amplification, such a test takes less than 2.5 hours to perform.

Example of patient follow-up showing the development of drug resistance as monitored by the INNO-LiPA HBV DR assay. For details, see text. Case history courtesy of HG Niesters (Dept. of Virology, University Medical Center, Rotterdam, The Netherlands).
The limitation of such hybridization-based methods (melting curve analysis, line probe assays) lies in their single-base discrimination. Specificity can be influenced by the sequences neighboring a polymorphic site, or by possible interference from secondary structures [36]. Furthermore, as new mutations arise, such hybridization-based assays must be updated accordingly.
Recent Developments
Possible future methods for detection of resistance could make use of technologies such as fluorescence (relying on secondary reporter systems) [37,38] or matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) [39,40]. They offer a highly sensitive means to detect unidentified HBV variants – even when comprising only a small percentage of wild-type/mutant mixtures. While fast and accurate, these methods require the use of well-trained technicians and expensive equipment, and have not been optimized for high-throughput use.
Oligonucleotide Microarrays
Microchip-based tests can also be envisaged for HBV resistance testing, as they may be well suited for the study of multiple mutations on the same genome [41]. However, as the number of clinically relevant mutants is presently relatively limited, the development of microarray-based assays for HBV resistance will probably not be cost effective unless combined with other tests.
(2) Indirect Methods
Quantification of HBV DNA (viral load assays)
Aside from its role as a 'baseline' indicator of HBV infectivity, the measurement of viral load is an indispensable means for monitoring and confirming resistance to lamivudine therapy. Indeed, resistance to antivirals is still generally defined clinically by a rise in serum HBV DNA levels during antiviral therapy.
Although HBV DNA assays will eventually detect changes in viral load as a consequence of emerging resistance to lamivudine (see further), other factors (e.g., compliance) may also be responsible for alterations in HBV DNA levels. Therefore, viral load measurements and changes in ALT titers should, by themselves, not be solely relied upon as indicators of viral resistance now that specific genotyping tests for drug resistance are available. Moreover, when a given mutation represents only a minor fraction of the total viral population, viral load will only be proportionately affected, and will rise with a variable delay with respect to the onset and progression of resistance (Figure 4). Therefore, the combined use of a genotyping assay for polymerase mutant detection and quantitative detection of viremia with a highly sensitive assay is warranted for optimal monitoring of HBV antiviral therapy [42,43].
(3) Phenotyping Methods
These methods detect drug resistance based on the use of molecular or cellular techniques or using animal models [20]. Such systems are useful for studying HBV replication, cellular accumulation of covalently closed circular DNA, and for characterizing HBV mutants. However, their use is labor-intensive and not amenable to high-throughput testing or commercialization.
6. Management of HBV Drug Resistance
Given the recent approval of adefovir dipivoxil, and the probability that other antivirals could also become available in the not-too-distant future, the management of drug resistance must now be seen within a broadening and evolving clinical context. Moreover, it is increasingly apparent that clinically relevant prediction and monitoring of viral resistance may not only require the use of existing tools such as drug resistance and viral load testing, but might also have to be extended to include the detection of mutations in the precore and core promoter regions as well as HBV genotyping. This is because recent studies have, for instance, shown that patients with precore mutants who develop lamivudine resistance may have a higher risk for severe exacerbation of liver disease [44,45]. This is especially worrying since chronic HBV patients harbouring precore mutants generally require long-term therapy. Numerous studies also indicate strong associations between certain viral genotypes that influence mutational patterns in the precore/core promoter regions responsible for either abolishing or diminishing HBeAg production [46,47].
7. Clinical Impact of Hepatitis B Genotypes
For a DNA virus, the HBV genome shows an exceptional degree of molecular variation (incorporation of some 1010 mistaken nucleotides into the HBV virion per day [21]). Over time, this spontaneous tendency for mutations has led to the emergence of at least seven HBV genotypes (designated A to G), defined by a divergence of 8% or more with reference to the complete nucleotide sequence. These different genotypes show a distinct geographic distribution: genotype A is found in northern Europe, the United States, and Central Africa; B and C predominate in Asia; D is associated with southern Europe, the Middle East, and India; E is uniquely African; and F is found in Central and South America as well as in Polynesia [48,49]. Genotype G has recently been localized in the United States and France [50], while a putative eighth genotype H has also been discovered in Central America [51]. The study of possible clinical ramifications of HBV genotypes is the subject of intense clinical research, with evidence accumulating that HBV genotyping influences the natural history and severity of liver disease, HBeAg seroconversion rates, precore/core promoter mutational patterns, and response to treatment (Table 3). Its determination – just as is the case for hepatitis C – could well emerge as an important consideration in the clinical evaluation of HBV patients prior to treatment.
Emerging clinical relevance of genotyping
| Characteristic | Country and Reference | Comment |
| HBeAg seroconversion | Hong Kong , Yuen et al. [52] | Earlier with genotype B than with C |
| Taiwan, Kao et al. [53] | Earlier with genotype B than with C | |
| Japan, Sumi et al. [54] | Earlier with genotype B than with C | |
| United States, Chu et al. [55] | Earlier with genotype B than with C | |
| Disease progression (natural history) | Hong Kong, Chan et al. [56] | More active liver disease with C than B |
| Taiwan, Kao et al. [57] | More severe disease with C incl. more HCC | |
| Japan, Duong et al. [58] | More chronic liver disease with C vs. asymptomatic with D + earlier seroconversion | |
| Japan, Sumi et al. [54] | Slower development of liver fibrosis and HCC with genotype B than with C | |
| India, Thakur et al. [59] | More severe liver disease with D than A | |
| Spain, Sanchez-Tapias et al. [60] | More deaths and severe disease outcome with F than with A or D | |
| Development of mutations | Hong Kong, Yuen et al. [61] | Rates to lamivudine same for B and C |
| Taiwan, Kao et al. [57] | Rates to lamivudine same for B and C | |
| Japan, Sumi et al. [54] | More core promoter mutants with C than B | |
| East Asia, Lindh et al. [46] | More core promoter mutants with C + more severe liver disease | |
| Response to interferon-alpha | Taiwan ,Kao et al. [57] | Better response with genotype B than C |
| Japan, Seo et al. [62] | Better response with genotype B than C | |
| U.S., Wai et al. [63] | More HBeAg clearance with B than C | |
| Response to lamivudine | Taiwan, Kao et al. [57] | Better virological response with B than C |
| Taiwan, Chien et al. [64] | More sustained response woth B than C |
8. Research Directions
Advances in HBV diagnostic testing have fortuitously paralleled the advent of new, effective drugs for HBV that have already been, or will soon be approved in the near future. These new assays - combined with classical diagnostic methods - will help determine the best long-term treatment strategies (sequential, combined, etc.) for the use of such new drugs. At the same time, a key challenge is the integration of HBV diagnostic testing with new therapeutic agents in order to ensure optimized, tailored patient treatment. Such integrated HBV diagnostic and treatment strategies will undoubtedly have to be adapted to economic imperatives and epidemiological realities (geographic patterns and modes of transmission). For instance, this implies differing testing-treatment schemes depending on whether high-incidence, less affluent regions or low-incidence, high-affluent areas are being targeted. Whatever the strategies that emerge, both classical HBV serological tests and advanced molecular diagnostics will be used together to monitor therapeutic efficacy and ensure cost-effectiveness.
Conflict of interest
Erwin Sablon and Fred Shapiro both work for Innogenetics NV.
References
1. Lok ASF, McMahon BJ. Chronic hepatitis B. Hepatology. 2001 ;34:1225-1241
2. Bowden S. Laboratory diagnosis of hepatitis B infection. In: (ed.) Lai CL, Locarnini S. Hepatitis B Virus. London, UK: International Medical Press. 2002:145-159
3. Hoofnagle JH, Dusheiko GM, Seef LB. et al. Seroconversion from hepatitis B e antigen to antibody in chronic type B hepatitis. Ann Intern Med. 1981 ;94:744-748
4. Davis GL. Update on the management of chronic hepatitis B. Rev Gastroenterol Disord. 2002 ;2:106-115
5. Schutten M, Niesters HGM. Clinical utility of viral quantification as a tool for disease monitoring. Exp Rev Mol Diagn. 2001 ;1:153-162
6. Saldanha J, Gerlich W, Lelie N, Dawson P, Heermann K, Heath A. An international collaborative study to establish a World Health Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques. Vox Sang. 2001 ;80:63-71
7. Conjeevaram HS, Lok ASF. Management of chronic hepatitis B. J Hepatol. 2003 ;38(suppl 1):S90-S103
8. Dienstag JL, Perrillo RP, Schiff ER, Bartholomew M, Vicary C, Rubin M. A preliminary trial of lamivudine for chronic hepatitis B infection. New Engl J Med. 1995 ;333:1657-1661
9. Lai CL, Ching CK, Tung AK, Li E, Young J, Hill A. et al. Lamivudine is effective in suppressing hepatitis B virus DNA in Chinese hepatitis B surface antigen carriers: a placebo-controlled trial. Hepatology. 1997 ;25:241-244
10. Cooksley WGE, Piratvisuth T, Lee S-D, Mahachai V, Chao T, Tanwandee T. et al. Peginterferon alpha-2a (40 kDa): an advance in the treatment of hepatitis B e antigen-positive chronic hepatitis B. J Viral Hepatitis. 2003 ;10:298-305
11. Lai C, Chien R, Leung N, Chang T, Guan R, Tai D. et al. A one-year trial of lamivudine for chronic hepatitis B. New Engl J Med. 1998 ;339:61-68
12. Dienstag JL, Schiff ER, Wright TL, Perrillo RP, Hann HW, Goodman Z. et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. New Engl J Med. 1999 ;341:256-1263
13. Lai C-L, Dienstag J, Schiff E, Leung NWY, Atkins M, Hunt C. et al. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis. 2003 ;36:687-696
14. Marcellin P, Chang TT, Lim SG, Tong MJ, Sievert W, Shiffman ML. et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N Engl J Med. 2003 ;348:808-816
15. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT, Kitis G, Rizzetto M. et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-negative chronic hepatitis B. N Engl J Med. 2003 ;348:800-807
16. Westland CE, Yang H, Delaney WE 4th, Gibbs CS, Miller MD, Wulfsohn M. et al. Week 48 resistance surveillance in two phase 3 clinical studies of adefovir dipivoxil for chronic hepatitis B. Hepatology. 2003 ;38:96-103
17. Yang H, Westland CE, Delaney WE 4th, Heathcote EJ, Ho V, Fry J. et al. Resistance surveillance in chronic hepatitis B patients treated with adefovir dipivoxil for up to 60 weeks. Hepatology. 2002 ;36:464-473
18. Angus P, Vaughan R, Xiong S, Yang H, Delaney W, Gibbs C. et al. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology. 2003 ;125:292-297
19. Delaney WE 4th, Locarnini S, Shaw T. Resistance of hepatitis B virus to antiviral drugs: current aspects and directions for future investigation. Antiviral Chem Chemother. 2001 ;12:1-35
20. Zoulim F. Assessing hepatitis B virus resistance in vitro and molecular mechanisms of nucleoside resistance. Semin Liver Dis. 2002 ;22:23-31
21. Doo E, Liang TJ. Molecular anatomy and pathophysiologic implications of drug resistance in hepatitis B virus infection. Gastroenterology. 2001 ;120:1000-1008
22. Nowak MA, Bonhoeffer S, Hill AM, Boehme R, Thomas HC, McDade H. Viral dynamics in hepatitis B virus infection. Proc Nat Acad Sci USA. 1996 ;93:4298-4402
23. Stuyver LJ, Locarnini SA, Lok A, Richman DD, Carman WF, Dienstag JL. et al. Nomenclature for antiviral-resistant human hepatitis B virus mutations in the polymerase region. Hepatology. 2001 ;33:751-757
24. Zoulim F. Detection of hepatitis B virus resistance to antivirals. J Clin Virol. 2001 ;21:243-253
25. Clarke B, Bloor S. Molecular genotyping of hepatitis B virus. J Clin Virol. 2002 ;25:S41-S45
26. Aberle SW, Kletzmayr J, Watschinger B, Schmeid B, Vetter N, Puchhammer-Stockl E. Comparison of sequence analysis and the INNO-LiPA HBV DR line probe assay for detection of lamivudine-resistant hepatitis B virus strains in patients under various clinical conditions. J Clin Microbiol. 2001 ;39:1972-1974
27. Stuyver L, Van Geyt C, De Gendt S, Van Reybroeck G, Zoulim F, Leroux-Roels G. et al. Line probe assay for monitoring drug resistance in hepatitis B virus-infected patients during antiviral therapy. J Clin Microbiol. 2000 ;38:702-707
28. Jardi R, Buti M, Rodriguez-Frias F, Cotrina M, Costa X, Pascual C. et al. Rapid detection of lamivudine-resistant hepatitis B virus polymerase gene variants. J Virol Meth. 1999 ;83:181-187
29. Allen MI, Gauthier J, DesLauriers M, Bourne EJ, Carrick KM, Baldanti F. et al. Two sensitive PCR-based methods for detection of hepatitis B variants associated with reduced susceptibility to lamivudine. J Clin Microbiol. 1999 ;37:3338-3347
30. Niesters HGM, De Man RA, Pas SD, Fries E, Osterhaus ADME. Identification of a new variant in the YMDD motif of the hepatitis B polymerase gene selected during lamivudine therapy. J Med Microbiol. 2002 ;51:695-699
31. Chayama K, Suzuki Y, Kobayashi M, Tsubota A, Hashimoto M, Miyano Y. et al. Emergence and takeover of YMDD motif mutant hepatitis B virus during long-term lamivudine therapy and re-takeover by wild type after cessation of therapy. Hepatology. 1998 ;27:1711-1716
32. Cane PA, Cook P, Ratcliffe D, Mutimer D, Pillay D. Use of real-time PCR and fluorimetry to detect lamivudine resistance-associated mutations in hepatitis B virus. Antimicrob Agents Chemother. 1999 ;43:1600-1608
33. Whalley SA, Brown D, Teo CG, Dusheiko GM, Saunders NA. Monitoring the emergence of hepatitis B virus polymerase gene variants during lamivudine therapy using the LightCycler. J Clin Microbiol. 2001 ;39:1456-1459
34. Punia P, Cane P, Teo C-G, Saunders N. Quantitation of hepatitis B lamivudine resistant mutants by real-time amplification refractory mutation system PCR. J Hepatol. 2004 ;40:986-992
35. Lok ASF, Zoulim F, Locarnini S, Mangia A, Niro G, Decraemer H. et al. Monitoring drug resistance in chronic hepatitis B virus (HBV)-infected patients during lamivudine therapy: evaluation of performance of INNO-LiPA HBV DR assay. J Clin Microbiol. 2002 ;40:3729-3734
36. Southern E, Mir K, Shchepinov M. Molecular interactions on microarrays. Nat Genet. 1999 ;21(suppl):5-9
37. Hall JG, Eis PS, Law SM, Reynaldo LP, Prudent JR, Marshall DJ. et al. Sensitive detection of DNA polymorphisms by the serial invasive signal amplification reaction. Proc Natl Acad Sci USA. 2000 ;97:8272-8277
38. Bai YJ, Zhao JR, Lv GT, Zhang WH, Wang Y, Yan XJ. Rapid and high throughput detection of HBV YMDD mutants with florescence polarization. World J Gastroenterol. 2003 ;9:2344-2347
39. Murray KK. DNA sequencing by mass spectrometry. J Mass Spectrom. 1996 ;31:1203-1215
40. Hong SP, Kim NK, Hwang SG, Chung HJ, Kim S, Han JH. et al. Detection of hepatitis B virus YMDD variants using mass spectrometric analysis of oligonucleotide fragments. J Hepatol. 2004 ;40:837-844
41. Wilson JW, Bean P, Robins T, Graziano F, Persing DH. Comparative evaluation of three human immunodeficiency virus genotyping systems: the HIV-GenotypR method, the HIV PRT GeneChip assay, and the HIV-RT line probe assay. J Clin Microbiol. 2000 ;38:3022-3028
42. Nafa S, Ahmed S, Tavan D, Pichoud C, Berby F, Stuyver L. et al. Early detection of viral resistance by determination of hepatitis B polymerase mutations in patients treated by lamivudine for chronic hepatitis B. Hepatology. 2000 ;32:1078-1088
43. Kirishima T, Okanoue T, Daimon Y, Itoh Y, Nakamura H, Morita A. et al. Detection of YMDD mutant using a novel sensitive method in chronic liver disease type B patients before and during lamivudine treatment. J Hepatol. 2002 ;37:259-265
44. Warner N, Bartholomeusz A, Edwards R, Vozzi V, Colledge D. et al. Functional analysis of HBV mutants selected after prolonged lamivudine treatment. Proceedings of the 11th International Symposium on Viral Hepatitis & Liver Disease (Abstract WE3-09). April 2003 Sydney Australia
45. Papatheodoridis GV, Dimou E, Laras A, Papadimitropoulos V, Hadziyannis SJ. Course of virologic breakthroughs under long-term lamivudine in HBeAg-negative precore mutant HBV liver disease. Hepatology. 2002 ;36:219-226
46. Lindh M, Hannoun C, Dhillon AP, Norkrans G, Horal P. Core promoter mutations and genotypes in relation to viral replication and liver damage in East Asian hepatitis B virus carriers. J Infect Dis. 1999 ;179:775-782
47. Grandjacques C, Pradat P, Stuyver L, Chavallier M, Chavallier P, Pichoud C. et al. Rapid detection of genotypes and mutations in the pre-core promoter and the pre-core region of hepatitis B virus genome: correlation with viral persistence and disease severity. J Hepatol. 2000 ;33:430-439
48. Okamoto H, Tsuda F, Sakugawa H, Sastrosoewignjo RI, Imai Mmiyakawa Y. et al. Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J Gen Virol. 1988 ;69:2575-2583
49. Magnius LO, Norder H. Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequence variability of the S-gene. Intervirology. 1995 ;38:24-34
50. Stuyver L, De Gendt S, Van Geyt C, Zoulim F, Fried M, Schinazi RF. et al. A new genotype of hepatitis B virus: complete genome and phylogenetic relatedness. J Gen Virol. 2000 ;81:67-74
51. Arauz-Ruiz P, Norder H, Robertson BH, Magnius LO. Genotype H: a new Amerindian genotype of hepatitis B virus revealed in Central America. J Gen Virol. 2002 ;83:2059-2073
52. Yuen MF, Sablon E, Tanaka Y, Kato T, Mizokami M, Doutreloigne J. et al. Epidemiological study of hepatitis B virus genotypes, core promoter and precore mutations of chronic hepatitis B infection in Hong Kong. J Hepatol. 2004 ;41:119-125
53. Kao JH, Chen PJ, Lai MY, Chen DS. Hepatitis B virus genotypes and spontaneous hepatitis B e antigen seroconversion in Taiwanese hepatitis B carriers. J Med Virol. 2004 ;72:363-369
54. Sumi H, Yokosuka O, Seki N, Arai M, Imazeki F, Kurihara T. et al. Influence of hepatitis B virus genotypes on the progression of chronic type B liver disease. Hepatology. 2003 ;37:19-26
55. Chu CJ, Hussain M, Lok AS. Hepatitis B virus genotype B is associated with earlier HBeAg seroconversion compared with hepatitis B virus genotype C. Gastroenterology. 2002 ;122:1756-1762
56. Chan HL, Wong ML, Hui AY Hung LC, Chan FK, Sung JJ. Hepatitis B virus genotype C takes a more aggressive disease course than hepatitis B virus genotype B in hepatitis B e antigen-positive patients. J Clin Microbiol. 2003 ;41:1277-1279
57. Kao JH. Hepatitis B virus genotypes and hepatocellular carcinoma in Taiwan. Intervirology. 2003 ;46:400-407
58. Duong TN, Horiike N, Michitaka K, Yan C, Mizokami M, Tanaka Y. et al. Comparison of genotypes C and D of the hepatitis B virus in Japan: a clinical and molecular biological study. J Med Virol. 2004 ;72:551-557
59. Thakur V, Guptan RC, Kazim SN, Malhotra V, Sarin SK. Profile, spectrum and significance of HBV genotypes in chronic liver disease patients in the Indian subcontinent. J Gastroenterol Hepatol. 2002 ;17:165-70
60. Sanchez-Tapias JM, Costa J, Mas A, Bruguera M, Rodes J. Influence of hepatitis B virus genotype on the long-term outcome of chronic hepatitis B in western patients. Gastroenterology. 2003 ;123:1848-1856
61. Yuen MF, Wong DK, Sablon E, Yuan HJ, Sum SM, Hui CK. et al. Hepatitis B virus genotypes B and C do not affect the antiviral response to lamivudine. Antivir Ther. 2003 ;8:531-534
62. Seo Y, Yoon S, Hamano K, Nakaji M, Yano Y, Katayama M. et al. Early response to interferon alpha treatment and long-term clinical outcome in Japanese patients with chronic HBV genotype C infection. Int J Mol Med. 2004 ;13:75-79
63. Wai CT, Chu CJ, Hussain M, Lok AS. HBV genotype B is associated with better response to interferon therapy in HBeAg(+) chronic hepatitis than genotype C. Hepatology. 2002 ;36:1425-1430
64. Chien RN Yeh CT, Tsai SL, Chu CM, Liaw YF. Determinants for sustained response to lamivudine therapy. Hepatology. 2003 ;38:1267-1273
Tables and Figures
Author biography
Erwin Sablon, PhD, is currently the Head of the Hepatology Unit, in the Diagnostics R&D Division at Innogenetics. His key interests include the molecular basis of drug resistance and genetic variability of the hepatitis B and C viruses, as well as serum markers for liver fibrosis.
Fred Shapiro, MLS, is the Head of the Medical Writing, Science Editing, and Science Information Departments at Innogenetics. His principle research interests include diagnosis and treatment of viral hepatitis, Alzheimer's disease, as well as the development of theranostics for personalized medicine.
![]() Corresponding address:
Corresponding address:
Erwin Sablon, PhD, Head of Hepatology Unit, Theranostics R&D, Innogenetics NV, Industriepark Zwijnaarde 7/4, B-9052 Gent, Belgium. Tel.:32-9-241-0804 Fax.: 32-9-241-0907Email: Erwin_Sabloncom