International Journal of Medical Sciences
ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2010; 7(2):72-81. doi:10.7150/ijms.7.72 This issue Cite
Research Paper
WT1 PEPTIDE VACCINATION IN COMBINATION WITH IMATINIB THERAPY FOR A PATIENT WITH CML IN THE CHRONIC PHASE
Miwako Narita1 ![]() , Masayoshi Masuko2, Tohri Kurasaki3, Toshiki Kitajima3, Shoko Takenouchi3, Anri Saitoh1, Norihiro Watanabe1, Tatsuo Furukawa2, Ken Toba3, Ichiro Fuse4, Yoshifusa Aizawa3, Manabu Kawakami5, Yoshihiro Oka6, Haruo Sugiyama6, Masuhiro Takahashi1
, Masayoshi Masuko2, Tohri Kurasaki3, Toshiki Kitajima3, Shoko Takenouchi3, Anri Saitoh1, Norihiro Watanabe1, Tatsuo Furukawa2, Ken Toba3, Ichiro Fuse4, Yoshifusa Aizawa3, Manabu Kawakami5, Yoshihiro Oka6, Haruo Sugiyama6, Masuhiro Takahashi1
1. Laboratory of Hematology and Oncology, Graduate School of Health Sciences, Niigata University, Niigata, Japan
2. Division of Stem Cell Transplantation, Niigata University Medical and Dental General Hospital, Niigata, Japan
3. Division of Hematology, Graduate School of Medical and Dental Sciences, Niigata University, Niigata, Japan
4. Division of Bioscience Medical Research Center, Niigata University Medical and Dental General Hospital, Niigata, Japan
5. Department of Medicine, National Hospital Organization, Osaka Minami Medical Center, Osaka, Japan
6. Department of Functional Diagnostic Science, Osaka University Graduate School of Medicine, Osaka, Japan
Received 2009-11-7; Accepted 2010-4-9; Published 2010-4-20
Citation:
Narita M, Masuko M, Kurasaki T, Kitajima T, Takenouchi S, Saitoh A, Watanabe N, Furukawa T, Toba K, Fuse I, Aizawa Y, Kawakami M, Oka Y, Sugiyama H, Takahashi M. WT1 PEPTIDE VACCINATION IN COMBINATION WITH IMATINIB THERAPY FOR A PATIENT WITH CML IN THE CHRONIC PHASE. Int J Med Sci 2010; 7(2):72-81. doi:10.7150/ijms.7.72. https://www.medsci.org/v07p0072.htm
Other stylesAbstract
Although tyrosine kinase inhibitors is effective for dramatically reducing CML cells, it might be difficult to eradicate completely the CML stem cells. We aimed to clarify the safety and effects of WT1 peptide vaccination in combination with imatinib therapy for a CML patient. A 51 year-old male with CML in CP, who showed a resistance against imatinib therapy for 2.5 years, began to be treated with 9mer modified-type WT1 peptides in combination with standard dose of imatinib. Although every 2-week-administration of WT1 peptides for 22 weeks did not show definite effects on the quantification of bcr-abl transcripts, by changing the administration from every 2 weeks to 4 weeks bcr-abl transcripts decreased remarkably. After 11 months of every 4-week-administration of the peptides and 12 months post cessation of the peptides bcr-abl transcripts achieved to the level below detection by RQ/RT-PCR (complete molecular response). WT1/MHC tetramer+CD8+ CTLs, which appeared after the second administration of WT1 peptides and remained more than 15 in number among 106 CD8+ T cells throughout the administration of WT1 peptides, are still present in the blood on 14th month post cessation of the peptides. An in vitro study as to the cytotoxicity of lymphocytes induced by mixed lymphocyte peptide culture demonstrated that cultured lymphocytes possessed cytotoxicity against WT1 expressing leukemia cells and the cytotoxicity was WT1-specific and MHC class I restricted. The present study showed that WT1 peptide vaccination in combination with TKI is feasible and effective in the therapy for imatinib-resistant CML.
Keywords: WT1 peptide vaccination, CML, imatinib, bcr-abl transcripts, WT1 tetramer, cytotoxicity
INTRODUCTION
While tyrosine kinase inhibitors (TKIs) such as imatinib are currently regarded as the first line therapy for chronic myelogenous leukemia (CML), it seems that CML stem cells display intrinsic resistance against most TKIs [1]. Therefore, extermination of CML stem cells could be critically needed for CML patients to be fully liberated from the TKI therapy. On the other hand, anti-tumor cytotoxic T lymphocytes (CTLs) are presumed to kill the relevant antigen-expressing tumor cells including resting cells such as tumor stem cells and spare normal hematopoietic progenitor cells [2].
Although the Wilms' tumor 1 (WT1) gene was first isolated as a tumor suppressor gene associated with the Wilms' tumor, the WT1 gene has been shown to be highly expressed in hematopoietic malignancies including acute and chronic myelogenous leukemia, acute lymphocytic leukemia and myelodysplastic syndrome, and a majority of solid tumors including glioblastoma, lung cancer, breast cancer, colorectal cancer, thyroid cancer, renal cancer, bone/soft tissue sarcoma and head/neck squamous cell carcinoma [3]. WT1-specific CTLs with HLA-A*0201 or A*2402 could be generated by stimulating peripheral blood mononuclear cells (PB-MNCs) with WT1 peptide-pulsed antigen presenting cells (APCs) in several laboratories [4-6]. WT1 peptides have already been used in clinical trials for specific immunotherapy of HLA-A24+ patients with brain tumor, breast cancer, colorectal cancer, thyroid cancer, leiomyosarcoma, or hematological malignancies including acute myeloid leukemia (AML), myeloma and myelodysplastic syndrome (MDS) [7].
In order to eradicate minimum residual CML cells which survived long-term imatinib therapy, we started WT1 peptide vaccination therapy in combination with imatinib in a CML patient who could not acquire a major molecular response through the administration with a single agent of imatinib. In addition, we tried to monitor the kinetics of WT1-specific CTLs as well as low frequency immunocompetent cells such as plasmacytoid DCs (pDCs), myeloid dendritic cell-1s (mDC1s), γδT cells and regulatory T (Treg) cells in peripheral blood (PB) during WT1 peptide vaccination.
MATERIALS AND METHODS
Administration of WT1 peptide
Modified-type WT1 peptide (HLA-A*2402-restricted, 9-mer peptide; CYTWNQMNL) was synthesized in GMP grade by NeoMPS (San Diego, CA, USA). WT1 peptide was dissolved with DMSO (Sigma-Aldrich, St. Louis, MO, USA) and then diluted with 5% glucose. A water-in-oil emulsion was prepared by mixing WT1 peptide as the aqueous phase and the adjuvant Montanide ISA-51 VG (Seppic, Paris, France) as the oil phase. The emulsion (WT1 peptide concentration: 3.33 mg/ml) was administered subcutaneously at the dose of 1 mg/body at 2 different sites such as the upper arm and thigh. The administration of WT1 peptides was performed after informed consent was obtained according to the protocol approved by the IRB of Niigata University School of Medicine.
Mixed lymphocyte peptide culture (MLPC)
MLPC was initiated by culturing 1-3 x105 PB-MNCs in 100 μl of 5% autologous serum-containing RPMI1640 with 10 μg modified-type WT1 peptides in 96 well plates in a modification of the method described by Karanikas et al [8]. Three days later RPMI1640 with 50 IU/ml IL-2 (Shionogi, Osaka, Japan) was added and a half of culture medium was changed every 3 days thereafter. After culturing for two weeks, cultured cells in each well were individually analyzed for various surface phenotypes, WT1 peptide/HLA-A*2402 tetramer and WT1-specific cytotoxicity by using flow cytometry.
Frequency of WT1 peptide/HLA-A*2402 tetramer+ cells
Modified-type WT1 peptide/HLA-A*2402 tetramer and HIV-1 env peptide (HLA-A*2402-restricted, 9-mer peptide; sequence: RYLRDQQLL)/HLA-A*2402 tetramer were kindly provided by Dr. K Kuzushima (Aichi Cancer Center Research Institute). Wild-type WT1 peptide (HLA-A*2402-restricted, modified 9-mer peptide; sequence: CMTWNQMNL)/HLA-A*2402 tetramer was purchased from MBL (Nagoya, Japan). For the tetramer assay, MLPC cells were double-stained with the FITC-CD8 antibody (BD Biosciences, San Jose, CA, USA) and PE-tetramer. HIV-1 env peptide/HLA-A*2402 tetramer was used as the negative control. Stained cells were analyzed with FACScan flow cytometry (BD Biosciences) and the data were analyzed by CellQest software (BD Biosciences). Frequency of WT1 peptide/HLA-A*2402 tetramer+ cells in PB-CD8+ cells was calculated by the following formula. Number of wells containing a lump of tetramer+CD8+ cells / (Number of PB-MNCs seeded in a well of MLPC) x (total number of wells for MLPC) x (ratio of number of PB-CD8+ cells in PB-MNCs). As to the frequencies of modified-type and wild-type WT1 peptide/HLA-A*2402 tetramer+CD8+ T cells, although the binding stability of wild-type WT1 peptide/HLA-A*2402 tetramer was lower than that of modified-type WT1 peptide/HLA-A*2402 tetramer, the frequencies of WT1 peptide/HLA-A*2402 tetramer+CD8+ T cells were almost the same between wild-type and modified-type tetramers. These data suggested that the frequency of wild-type WT1 peptide/ HLA-A*2402 tetramer+CD8+ T cells could be replaced by that of modified-type WT1 peptide/ HLA-A*2402 tetramer+CD8+ T cells. Therefore, in the present study, the frequency of WT1/MHC tetramer+CD8+ T cells was calculated from flow cytometry analysis using modified-type WT1 peptide/HLA-A*2402 tetramer.
Identification of pDCs, mDC1s, γδT cells, Treg cells
For identification of pDCs, mDC1s, γδT cells and Treg cells, PB-mononuclear cells (MNCs) were prepared from a WT1 peptide-treated patient and stained with FITC, PE or APC-labeled various monoclonal antibodies. pDCs were identified as CD303+ (BDCA2; Miltenyi Biotec, Bergisch Gladbach, Germany) cells. mDC1s were identified as lineage (CD3, CD14, CD19 and CD56)- / CD1c+ (BDCA1; Miltenyi Biotec) cells. γδT cells were identified as CD3+/γδTCR+ cells. For identification of Treg cells, PB-MNCs were stained with surface molecules such as CD4 and CD25 (BD Biosciences). Then the cells were treated with freshly prepared Fixation/Permeabilization working solution (eBioscience, San Diego, CA) for 60 minutes and stained with PE-conjugated anti-human Foxp3 antibody (eBioscience) or isotype control. Treg cells were identified as CD4+/Foxp3+ cells. Cell fluorescence was analyzed with FACSAria flow cytometry (BD Biosciences) and 10,000 events were collected, the data of which was analyzed by FACSDiva software (BD Biosciences).
Cytotoxicity assay
To estimate anti-WT1 cytotoxicity of MLPC cells, a 5,6-carboxy-fluorescein succinimidyl ester (CFSE; Molecular Probes, Eugene, OR)-based cytotoxicity assay was performed. Autologous EB-virus transformed B-lymphoblastoid cell line (B-LCL) cells pulsed with WT1 peptides were labeled with 10 μM CFSE, and were used as target cells for the cytotoxicity assay. Labeled target cells were co-cultured in tubes with effector cells for 4 hours at 37ºC in a fully humidified 5% CO2 atmosphere. Co-cultured cells (consisting of effector cells and target cells) were stained with 7AAD to identify dead cells, and a fixed amount (10,000 beads/tube) of FITC-labeled CaliBRITE beads (BD Biosciences) were added for quantitative analysis of the cell population just prior to flow cytometry analysis. Viable target cells (CFSE+/7AAD-) and CaliBRITE beads were gated in FSC/SSC and FL-1/FSC dot plots respectively in target cells, which had been cultured without effector cells. For each sample tube containing target cells with effector cells at various effector-to-target ratios, 5,000 CaliBRITE beads were acquired, which made it possible to calculate the absolute numbers of viable target cells. Percent cytotoxicity of the assay was calculated by the following formula. : % cytotoxicity = [(absolute number of viable target cells in the tube containing target cells only -absolute number of viable target cells in the sample tube containing target cells and effector cells)/absolute number of viable target cells in the tube containing target cells only] x 100.
Unlabeled target cell-mediated blocking of cytotoxicity by CTLs
CFSE-labeled target cells were mixed with the same unlabeled target cells at the ratio of 1:5-20 (CSFE-labeled target cells : unlabeled target cells) and co-cultured with effector cells for the cytotoxicity assay.
Anti-MHC class I monoclonal antibody-mediated blocking of cytotoxicity by CTLs
Target cells were incubated with anti-MHC class I monoclonal antibody (clone W6/32, mouse IgG2a; Serotec, Oxford, UK), anti-MHC class II monoclonal antibody (clone Tu39, mouse IgG2a; BD Pharmingen, San Diego, CA) or isotype control (clone MPC-11, mouse IgG2a; BD Pharmingen) at 10 μg/ml for 30 min and then co-cultured with effector cells for the cytotoxicity assay.
RESULTS
1. Clinical efficacy of WT1 peptide vaccination
A 51 year-old male with CML in chronic phase had been treated with 400 mg imatinib for two and a half years. Although bcr-abl transcripts decreased transiently to less than 1,000 copies in 1 μg RNA extracted from PB cells (3-log reduction = 280 copies in 1 μg cellular RNA; median in our laboratory, n=120) during the imatinib treatment, the transcripts gradually increased to more than 4,000 copies spontaneously thereafter. Imatinib was increased to a dose of 600 mg and continued for 4 months, which caused adverse effects such as worsening of anemia and limb pain with increased CK. Therefore the dose of imatinib was decreased to 400 mg. Thereafter bcr-abl transcripts decreased transiently to 500 copies during the imatinib treatment, which was speculated to be the late effects of imatinib therapy at the dose of 600 mg a day. However, bcr-abl transcripts gradually increased to more than 1,000 copies thereafter. Since the patient was HLA-A*2402+ and informed consent was obtained, modified-type WT1 peptides, which had been identified to possess an anti-tumor immunogenicity [7], were administered subcutaneously at the dose of 1 mg every 2 weeks in combination with 400 mg imatinib. After the second administration of the peptides, WT1 peptide/HLA-A*2402 (WT1/MHC) tetramer+CD8+ T cells began to be detected in PB at a frequency of 7x10-6 in PB-CD8+ T cells. After the fourth administration of WT1 peptides, bcr-abl transcripts decreased to 820 copies with an increase in the frequency of WT1/MHC tetramer+CD8+ T cells in PB. However, bcr-abl transcripts increased to more than 1,000 copies after the eighth administration of WT1 peptides, though the frequencies of WT1/MHC tetramer+CD8+ T cells in PB were maintained at a considerable level. Since there was a report that some anti-tumor CD8+ CTLs lose their cytolytic activity by strong antigenic stimulation [9], the interval of WT1 peptide administration was changed from two weeks to four weeks after the eleventh administration of WT1 peptides. Although bcr-abl transcripts rose up to 2,600 copies after 12th administration of WT1 peptides, thereafter the transcripts tended to decrease and fell to 400 copies after 22nd administration of WT1 peptides. After seven months from the cessation of WT1 peptide vaccination bcr-abl transcripts decreased to the level of a major molecular response (170 copies). Four months thereafter, bcr-abl transcripts achieved to the level below detection by RQ/RT-PCR (complete molecular response) (Fig. 1). WT1/MHC tetramer+CD8+ CTLs are still present in the blood on 14th month post cessation of the peptides. No adverse effects due to WT1 peptide vaccination was observed except for skin induration and redness at the sites of WT1 peptide injection.
2. Frequency of WT1/MHC tetramer+CD8+ T cells
The amplification of WT1-specific CTLs in PB by WT1 peptide vaccination was confirmed by evaluating the frequency of WT1/MHC tetramer+CD8+ T cells by using MLPC. Although WT1/MHC tetramer+CD8+ T cells were not detected in PB before the WT1 peptide vaccination, WT1/MHC tetramer+CD8+ T cells appeared after the second vaccination. By repeating the WT1 peptide vaccination, the frequency of WT1/MHC tetramer+CD8+ T cells was elevated to more than 15x10-6 in CD8+ T cells (Fig. 2).
Figure 1
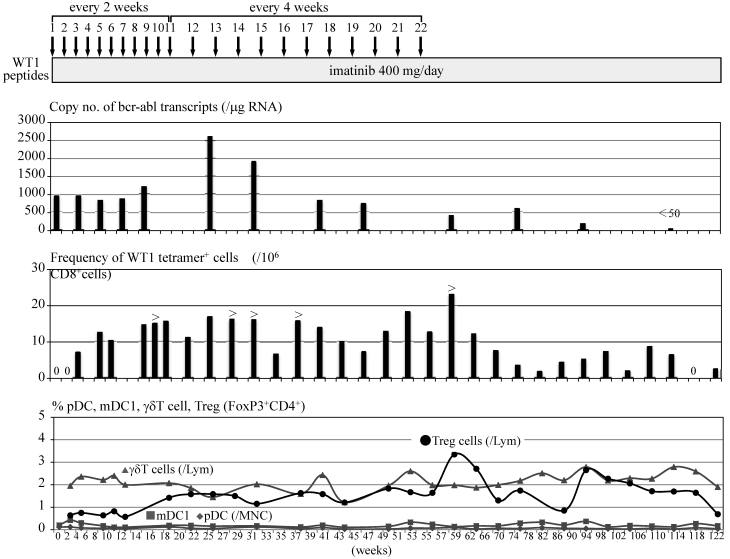
Clinical course of a CML patient treated with WT1 peptide vaccination. Figure shows copy numbers of bcr-abl transcripts/μg RNA extracted from PB-nucleated cells, frequency of WT1/MHC tetramer+CD8+ cells in 106 PB-CD8+ cells and percentage of PB-low frequent immunocompetent cells such as pDCs, mDC1s, γδT cells and Treg cells.
Figure 2
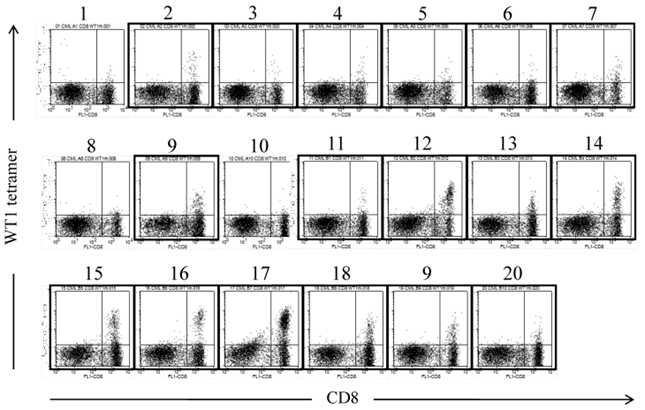
WT1 peptide/MHC tetramer analysis of MLPC cells cultured with WT1 peptides and IL-2 for 14 days. MLPC was performed in a 200 μl medium-containing well by culturing 1-3x105 MNCs of PB drawn from a CML patient at 4 weeks after the 20th administration of the WT1 peptide vaccination. MLPC cells in 20 wells (No. 1-20) were stained with FITC-CD8 and PE-modified-type WT1 peptide/MHC tetramer. Framed dot plots represent MLPC cells, which were evaluated as positive for WT1 peptide/MHC tetramer+CD8+ T cells. There were no MHC tetramer+CD8+ T cells in MLPC cells stained with HIV peptide/MHC tetramer.
3. Kinetics of immunocompetent cells
Complete blood cell counts including white blood cell differentials did not change before or after a WT1 peptide vaccination was added to imatinib therapy. In addition, routine flow cytometry examinations showed that lymphocyte subpopulations such as T cells, B cells, NK cells and NKT cells did not change during the course. Minor populations of immunocompetent cells such as pDCs, mDC1s and γδT cells and Treg cells were analyzed repeatedly during the WT1 peptide vaccination. Although there was no change observed in the numbers of pDCs, mDC1s and γδT cells, Treg cells increased after 8-9th WT1 peptide vaccination from less than 1% of all lymphocytes to nearly 2-3 % with a peak at the termination of WT1 peptide vaccination. Treg cells tended to decrease after cessation of WT1 peptide vaccination (Fig. 2).
4. Cytotoxicity of MLPC cells
The cytotoxic ability of cells grown by MLPC in each well was evaluated using the CFSE-labeled autologous B-LCL pulsed with modified or wild-type WT1 peptides. Cells in almost all the wells demonstrated a remarkable cytotoxicity against modified-type WT1 peptide-pulsed B-LCL. Although the level of cytotoxicity against wild-type WT1 peptide-pulsed B-LCL was less than that against modified-type WT1 peptide-pulsed B-LCL, cells grown in most wells of MLPC showed definite cytotoxicity against autologous wild-type WT1 peptide-pulsed B-LCL (Fig. 3). The cytotoxicity of MLPC cells against CFSE-labeled autologous B-LCL pulsed with WT1 peptides was blocked remarkably by adding CFSE-unlabeled target cells in a cold inhibition test. The cytotoxic ability of MLPC cells was blocked remarkably by adding antibodies against MHC class I in a cytotoxicity test using WT1 peptide-pulsed autologous B-LCL as target cells, but not by adding antibodies against MHC class II (Fig. 4).
MLPC cells in wells with WT1/MHC tetramer+CD8+ T cells showed cytotoxicity also against the HLA-A*2402+ leukemia cell line (C2F8) expressing WT1 intrinsically (Fig. 5).
Figure 3
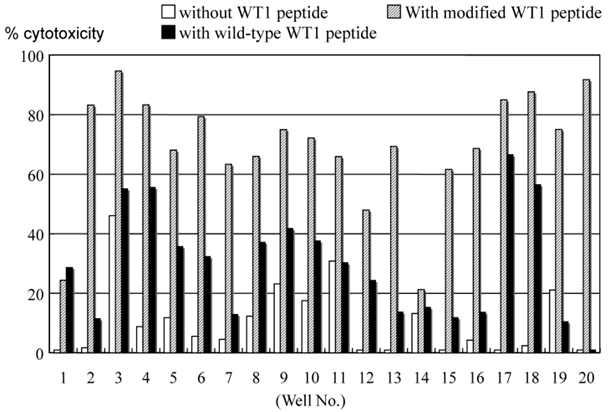
Cytotoxicity assay of cells cultured in each well of MLPC for 2 weeks. Autologous B-LCL pulsed with or without modified or wild-type WT1 peptides were used as target cells in a CFSE-labeled target cytotoxicity assay of effector : target ratio of 5:1.
Figure 4
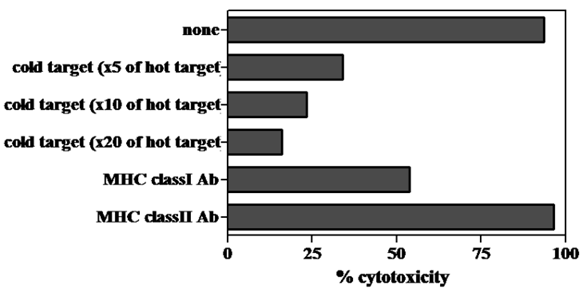
Unlabeled target cell- or anti-MHC class I monoclonal antibody-mediated blocking of cytotoxicity by MLPC cells. A CFSE-labeled target cell cytotoxicity assay was performed by using MLPC cells as effector cells and autologous B-LCL pulsed with modified-type WT1 peptides as target cells. CFSE-labeled B-LCL were mixed with 5-20 times unlabeled B-LCL (cold target) and co-cultured with MLPC cells at effector : the CFSE-labeled target ratio of 3:1 for the cold target inhibition test. The same CFSE-labeled target cells were incubated with anti-MHC class I or II monoclonal antibodies at 10 μg/ml for 30 min and then co-cultured with MLPC cells at effector : target ratio of 3:1 for the anti-MHC class I monoclonal antibody inhibition test. This is a representative experiment among four similar experiments. The other three similar experiments induced the identical results as one inserted in Figure 5.
Figure 5
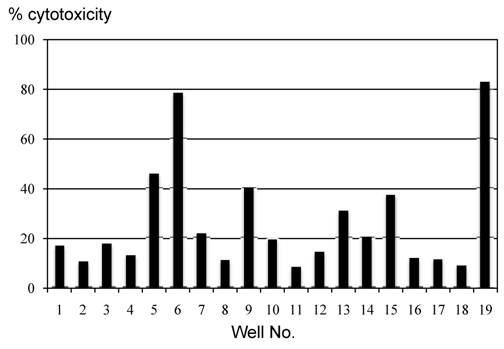
Cytotoxicity assay of MLPC cells against leukemia cells expressing WT1 intrinsically. A CFSE-labeled target cytotoxicity assay was performed by using cells cultured in each well of MLPC for 2 weeks as effector cells and a CML blastic crisis cell line expressing HLA-A*2402, C2F8, as target cells at effector : target ratio of 5:1. MLPC was initiated by using MNCs of PB drawn from a CML patient at 3 weeks after the 2nd administration of WT1 peptide vaccination.
DISCUSSION
Antigenic Peptide derived from leukemia-associated antigens such as WT1 protein, proteinase-3, receptor for hyaluronic acid-mediated motility (RHAMM) and bcr-abl fusion protein have been demonstrated to induce antigen-specific CTLs against leukemia cells in preclinical studies [10-13]. Preliminary clinical trials using peptide vaccines of these leukemia-associated antigens have shown that the clinical response is associated with generation of antigen-specific CTLs in patients with CML, AML and MDS [14-23]. In the peptide vaccination for patients with CML, Bocchia et al showed further reduction of residual disease with a complete molecular response in CML patients undergoing imatinib treatment by vaccinating six times with bcr-abl-derived peptides plus GM-CSF and GS-21 as adjuvant [17]. Thereafter they reported a longer follow up study suggesting that a 6 month interval between bcr-abl vaccination boosts was too long to maintain an efficient immune control on residual leukemic cells [24]. Rojas et al administered HLA class I-binding bcr-abl peptides alone or with the pan HLA DR-binding epitope (PADRE) to augment CD4+ T cells and reported that the development of an anti-bcr-abl T cell response correlated with a subsequent fall in bcr-abl transcripts [16]. Maslak et al performed a clinical trial administering bcr-abl fusion peptides including modified-type bcr-abl peptides eleven times: five doses biweekly, four doses monthly, and then at 9 and 12 months. Although a T cell response against natural-type bcr-abl peptides was demonstrated even in CML patients treated with modified-type bcr-abl fusion peptides, they could not show that the T cells were capable of killing fresh CML cells or that there were any clear clinical responses that correlated with the immune responses [25]. As to WT1 peptide vaccination for patients with leukemia, WT1 peptide vaccination for AML [20, 26], overt leukemia from MDS, and MDS with myelofibrosis [27] have been previously reported. Kawakami et al reported that vaccination of a secondary chronic myelomonocytic leukemia patient with very low-dose (5 μg/body) of WT1 peptides could generate a WT1-specific CTL response and lead to a reduction of the WT1 transcript level [21]. Recently Razvani et al reported a clinical trial using a combination of PR1 peptides (peptides derived from proteinase 3) and WT1 peptides in patients with myeloid malignancies including AML in complete remission, MDS and CML in the chronic phase. They demonstrated that the post-vaccination emergence of PR1 or WT1-specific CD8+ T cells was associated with a decrease in WT1 mRNA expression. The vaccination consisted of just one subcutaneous administration of the peptides and one CML patient showed a minimal response to the combined PR1 and WT1 peptide vaccination [28]. Qazilbash et al reported vaccination with PR1 peptides and GM-CSF in 10 patients with CML who did not respond to upfront treatment or who experienced relapse of the disease. They showed that 1 patient had cytogenetic CR and three patients refractory to allogeneic transplantation, interferon and imatinib had stable disease with some hematological improvement [22].
We administered WT1 peptides to one patient who showed an increase of bcr-abl transcripts after getting into complete cytogenetic response but not 3-log reduction throughout the previous treatment with imatinib for two and a half years. Modified-type WT1 peptides were used instead of wild-type WT1 peptides because modified-type WT1 peptides had been demonstrated to possess a much higher binding capacity to the complementary determining region (CDR) of HLA-A*2402 and a more potent capacity to generate WT1 specific CTLs in vitro than wild-type WT1 peptides. By administration of WT1 peptides every two weeks in combination with the same amount of imatinib, the tendency of increase of bcr-abl transcripts was abrogated and the patient showed a slight decrease of bcr-abl transcripts after the 4th administration of WT1 peptides. However, by repeated every two-week administration of WT1 peptides, bcr-abl transcripts tended to increase after the 8th administration of WT1 peptides. Referring to a report that the tetramer binding capacity and specific cytotoxicity of the CTL line decreases remarkably by the stimulation of relevant peptide-pulsed cells [9], we performed an in vitro study regarding the effects of WT1 peptide stimulation on the kinetics of WT1/MHC tetramer+CD8+ cells in cells generated by MLPC for 2 weeks. Our study demonstrated that stimulation with WT1 peptides promptly decreased the absolute number of WT1/MHC tetramer+CD8+ cells and the decrease was maintained for two to three weeks. On the other hand, in MLPC cells without the addition of WT1 peptides, the absolute number of WT1/MHC tetramer+CD8+ cells increased and maintained a high level during the entire period of the culturing (data not shown). Considering that the same phenomenon could occur in vivo, we changed WT1 peptide administration from every two weeks to every four weeks from the 12th administration. Although bcr-abl transcripts increased considerably after the 12th administration of WT1 peptides, there was a gradual decrease of the transcripts thereafter to the level of complete molecular response after 11 months from the cessation of WT1 peptide administdration for 17 months (total administration: 22 times, every four-week administration: 11 times).
As to the frequency of WT1/MHC tetramer+CD8+ cells in PB of the patients treated with WT1 peptides, WT1 tetramer+CD8+ cells, which were not detected before the administration of WT1 peptides, appeared after the second administration of WT1 peptides. Even in the period in which bcr-abl transcripts kept increasing from 16 to 29 weeks of WT1 peptide treatment, the frequency of WT1/MHC tetramer+CD8+ cells maintained a high level. This may be due to blood drawn two or four weeks after the administration of WT1 peptides (immediately before the next administration). This is the time in which WT1/MHC tetramer+CD8+ cells had recovered from the nadir phase caused by WT1 peptides administered two or four weeks previously. It is speculated that during this period (at least for two weeks after the administration of WT1 peptides), the number of WT1/MHC tetramer+CD8+ cells had decreased and the cytotoxicity of anti-WT1 CTLs had been low, which could be associated with an elevation of bcr-abl transcripts.
Cells induced by MLPC for two weeks showed cytotoxicity against the leukemia cell line expressing both HLA-A*2402 and WT1 antigen as well as WT1 peptide-pulsed B-LCL. The cytotoxicity of MLPC cells was suppressed by adding non-labeled target cells in the cytotoxicity assay using CFSE-labeled and WT1 peptide pulsed autologous B-LCL as target cells. Besides, the cytotoxicity of MLPC cells was suppressed by treating target cells with anti-MHC class I monoclonal antibodies but not with anti-MHC class II monoclonal antibodies in the CFSE-labeled target cytotoxicity assay. These findings revealed that MLPC cells possess cytotoxicity specific to the WT1 antigen and restricted to MHC class I.
By repeated administration of WT1 peptides, Treg cells increased after 20 weeks from the initiation of WT1 vaccination, which was a simultaneous event as increase of bcr-abl transcripts of PB cells. The association of an increase of Treg cells and elevation of bcr-abl transcripts is not clear but it can be easily speculated that Treg cells might play a negative role in anti-tumor immunotherapy as in this case. These data suggested the possible usefulness of treatment for suppressing the function of regulatory T cells in combination with anti-tumor immunotherapy including tumor antigen peptide vaccination.
The present study showed that WT1 peptide vaccination for imatinib-pretreated CML patients is feasible and presumed to be effective for reducing leukemia cells, which was supported by the appearance of WT1/MHC tetramer+CD8+ T cells with anti-leukemic cytotoxicity in PB of a CML patient treated with WT1 peptide vaccination.
Acknowledgements
We greatly appreciate Dr. Kuzushima K (Aichi Cancer Center Research Institute) for his generous donation of modified-type WT1 peptide/HLA-A*2402 tetramer and HIV-1 env peptide/HLA-A*2402 tetramer. We thank also Dr. Suzuki S (T Cell Technologies Co., LTD) for his giving us valuable instructions in MLPC.
CONFLICT OF INTEREST
The authors declare that no conflict of interest exists.
References
1. Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, Eaves C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926-935
2. Gao L, Bellantuono I, Elsasser A, Marley SB, Gordon MY, Goldman JM, Stauss HJ. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood. 2000;95:2198-2203
3. Oka Y, Tsuboi A, Kawakami M, Elisseeva OA, Nakajima H, Udaka K, Kawase I, Oji Y, Sugiyama H. Development of WT1 peptide cancer vaccine against hematopoietic malignancies and solid cancers. Curr Med Chem. 2006;13:2345-2352
4. Oka Y, Elisseeva OA, Tsuboi A, Ogawa H, Tamaki H, Li H, Oji Y, Kim EH, Soma T, Asada M, Ueda K, Maruya E, Saji H, Kishimoto T, Udaka K, Sugiyama H. Human cytotoxic T-lymphocyte responses specific for peptides of the wild-type Wilms' tumor gene (WT1) product. Immunogenetics. 2000;51:99-107
5. Bellantuono I, Gao L, Parry S, Marley S, Dazzi F, Apperley J, Goldman JM, Stauss HJ. Two distinct HLA-A0201-presented epitopes of the Wilms tumor antigen 1 can function as targets for leukemia-reactive CTL. Blood. 2002;100:3835-3837
6. Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood. 2000;95:286-293
7. Oka Y, Tsuboi A, Oji Y, Kawase I, Sugiyama H. WT1 peptide vaccine for the treatment of cancer. Curr Opin Immunol. 2008;20:211-220
8. Karanikas V, Lurquin C, Colau D, van Baren N, De Smet C, Lethe B, Connerotte T, Corbiere V, Demoitie MA, Lienard D, Dreno B, Velu T, Boon T, Coulie PG. Monoclonal anti-MAGE-3 CTL responses in melanoma patients displaying tumor regression after vaccination with a recombinant canarypox virus. J Immunol. 2003;171:4898-4904
9. Demotte N, Stroobant V, Courtoy PJ, Van Der Smissen P, Colau D, Luescher IF, Hivroz C, Nicaise J, Squifflet JL, Mourad M, Godelaine D, Boon T, van der Bruggen P. Restoring the association of the T cell receptor with CD8 reverses anergy in human tumor-infiltrating lymphocytes. Immunity. 2008;28:414-424
10. Bocchia M, Korontsvit T, Xu Q, Mackinnon S, Yang SY, Sette A, Scheinberg DA. Specific human cellular immunity to bcr-abl oncogene-derived peptides. Blood. 1996;87:3587-3592
11. Molldrem J, Dermime S, Parker K, Jiang YZ, Mavroudis D, Hensel N, Fukushima P, Barrett AJ. Targeted T-cell therapy for human leukemia: cytotoxic T lymphocytes specific for a peptide derived from proteinase 3 preferentially lyse human myeloid leukemia cells. Blood. 1996;88:2450-2457
12. Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, Jones C, Housman DE. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell. 1990;60:509-520
13. Chen J, Schmitt A, Bunjes D, Chen B, Schmitt M. The receptor for hyaluronic acid-mediated motility induces specific CD8+ T cell response in healthy donors and patients with chronic myeloid leukemia after allogeneic stem cell transplantation. Int J Oncol. 2007;30:1119-1127
14. Pinilla-Ibarz J, Cathcart K, Korontsvit T, Soignet S, Bocchia M, Caggiano J, Lai L, Jimenez J, Kolitz J, Scheinberg DA. Vaccination of patients with chronic myelogenous leukemia with bcr-abl oncogene breakpoint fusion peptides generates specific immune responses. Blood. 2000;95:1781-1787
15. Cathcart K, Pinilla-Ibarz J, Korontsvit T, Schwartz J, Zakhaleva V, Papadopoulos EB, Scheinberg DA. A multivalent bcr-abl fusion peptide vaccination trial in patients with chronic myeloid leukemia. Blood. 2004;103:1037-1042
16. Rojas JM, Knight K, Wang L, Clark RE. Clinical evaluation of BCR-ABL peptide immunisation in chronic myeloid leukaemia: results of the EPIC study. Leukemia. 2007;21:2287-2295
17. Bocchia M, Gentili S, Abruzzese E, Fanelli A, Iuliano F, Tabilio A, Amabile M, Forconi F, Gozzetti A, Raspadori D, Amadori S, Lauria F. Effect of a p210 multipeptide vaccine associated with imatinib or interferon in patients with chronic myeloid leukaemia and persistent residual disease: a multicentre observational trial. Lancet. 2005;365:657-662
18. Mailander V, Scheibenbogen C, Thiel E, Letsch A, Blau IW, Keilholz U. Complete remission in a patient with recurrent acute myeloid leukemia induced by vaccination with WT1 peptide in the absence of hematological or renal toxicity. Leukemia. 2004;18:165-166
19. Keilholz U, Menssen HD, Gaiger A, Menke A, Oji Y, Oka Y, Scheibenbogen C, Stauss H, Thiel E, Sugiyama H. Wilms' tumour gene 1 (WT1) in human neoplasia. Leukemia. 2005;19:1318-1323
20. Oka Y, Tsuboi A, Taguchi T, Osaki T, Kyo T, Nakajima H, Elisseeva OA, Oji Y, Kawakami M, Ikegame K, Hosen N, Yoshihara S, Wu F, Fujiki F, Murakami M, Masuda T, Nishida S, Shirakata T, Nakatsuka S, Sasaki A, Udaka K, Dohy H, Aozasa K, Noguchi S, Kawase I, Sugiyama H. Induction of WT1 (Wilms' tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc Natl Acad Sci U S A. 2004;101:13885-13890
21. Kawakami M, Oka Y, Tsuboi A, Harada Y, Elisseeva OA, Furukawa Y, Tsukaguchi M, Shirakata T, Nishida S, Nakajima H, Morita S, Sakamoto J, Kawase I, Oji Y, Sugiyama H. Clinical and immunologic responses to very low-dose vaccination with WT1 peptide (5 microg/body) in a patient with chronic myelomonocytic leukemia. Int J Hematol. 2007;85:426-429
22. Qazilbash MH, Wieder E, Rios R, Lu S, Kant S, Giralt S, Estey EH, Thall P, de Lima M, Couriel D, Champlin RE, Komanduri K, Molldrem JJ. Vaccination with the PR1 leukemia-associated antigen can induce complete remission in patients with myeloid leukemia. Blood. 2004;104:259a
23. Schmitt M, Schmitt A, Rojewski MT, Chen J, Giannopoulos K, Fei F, Yu Y, Gotz M, Heyduk M, Ritter G, Speiser DE, Gnjatic S, Guillaume P, Ringhoffer M, Schlenk RF, Liebisch P, Bunjes D, Shiku H, Dohner H, Greiner J. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood. 2008;111:1357-1365
24. Bocchia M, Abruzzese E, Ippoliti M, Pirrotta M, Trawinska M, Amabile M, Martinelli G, Forconi F, Gozzetti A, Carella A, Raspadori D, Lauria F. Control of Residual Disease in Imatinib Treated Chronic Myeloid Leukemia Patients with Peptide Vaccinations: 2 Years Follow up of CMLVAX100 Trial. Blood. 2005;106:167a
25. Maslak PG, Dao T, Gomez M, Chanel S, Packin J, Korontsvit T, Zakhaleva V, Pinilla-Ibarz J, Berman E, Scheinberg DA. A pilot vaccination trial of synthetic analog peptides derived from the BCR-ABL breakpoints in CML patients with minimal disease. Leukemia. 2008;22:1613-1616
26. Keilholz U, Scheibenbogen C, Letsch A, Asemissen A, Hofmann W, Uharek L, Wolfgang B, Eckhard E. WT1-Peptide Vaccination Shows High Immunogenicity and Clinical Activity in Patients with Acute Myeloid Leukemia. Blood. 2005;106:406a
27. Oka Y, Tsuboi A, Murakami M, Hirai M, Tominaga N, Nakajima H, Elisseeva OA, Masuda T, Nakano A, Kawakami M, Oji Y, Ikegame K, Hosen N, Udaka K, Yasukawa M, Ogawa H, Kawase I, Sugiyama H. Wilms tumor gene peptide-based immunotherapy for patients with overt leukemia from myelodysplastic syndrome (MDS) or MDS with myelofibrosis. Int J Hematol. 2003;78:56-61
28. Rezvani K, Yong AS, Mielke S, Savani BN, Musse L, Superata J, Jafarpour B, Boss C, Barrett AJ. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood. 2008;111:236-242
Author contact
![]() Corresponding author: Miwako Narita M.D., Laboratory of Hematology and Oncology, Graduate School of Health Sciences, Niigata University, 2-746, Asahimachi-dori, Chuo-ku, Niigata, 951-8518 Japan. (Telephone/fax) 81-25227-0836, (email) naritaminiigata-u.ac.jp
Corresponding author: Miwako Narita M.D., Laboratory of Hematology and Oncology, Graduate School of Health Sciences, Niigata University, 2-746, Asahimachi-dori, Chuo-ku, Niigata, 951-8518 Japan. (Telephone/fax) 81-25227-0836, (email) naritaminiigata-u.ac.jp
Citation styles
APA
Narita, M., Masuko, M., Kurasaki, T., Kitajima, T., Takenouchi, S., Saitoh, A., Watanabe, N., Furukawa, T., Toba, K., Fuse, I., Aizawa, Y., Kawakami, M., Oka, Y., Sugiyama, H., Takahashi, M. (2010). WT1 PEPTIDE VACCINATION IN COMBINATION WITH IMATINIB THERAPY FOR A PATIENT WITH CML IN THE CHRONIC PHASE. International Journal of Medical Sciences, 7(2), 72-81. https://doi.org/10.7150/ijms.7.72.
ACS
Narita, M.; Masuko, M.; Kurasaki, T.; Kitajima, T.; Takenouchi, S.; Saitoh, A.; Watanabe, N.; Furukawa, T.; Toba, K.; Fuse, I.; Aizawa, Y.; Kawakami, M.; Oka, Y.; Sugiyama, H.; Takahashi, M. WT1 PEPTIDE VACCINATION IN COMBINATION WITH IMATINIB THERAPY FOR A PATIENT WITH CML IN THE CHRONIC PHASE. Int. J. Med. Sci. 2010, 7 (2), 72-81. DOI: 10.7150/ijms.7.72.
NLM
Narita M, Masuko M, Kurasaki T, Kitajima T, Takenouchi S, Saitoh A, Watanabe N, Furukawa T, Toba K, Fuse I, Aizawa Y, Kawakami M, Oka Y, Sugiyama H, Takahashi M. WT1 PEPTIDE VACCINATION IN COMBINATION WITH IMATINIB THERAPY FOR A PATIENT WITH CML IN THE CHRONIC PHASE. Int J Med Sci 2010; 7(2):72-81. doi:10.7150/ijms.7.72. https://www.medsci.org/v07p0072.htm
CSE
Narita M, Masuko M, Kurasaki T, Kitajima T, Takenouchi S, Saitoh A, Watanabe N, Furukawa T, Toba K, Fuse I, Aizawa Y, Kawakami M, Oka Y, Sugiyama H, Takahashi M. 2010. WT1 PEPTIDE VACCINATION IN COMBINATION WITH IMATINIB THERAPY FOR A PATIENT WITH CML IN THE CHRONIC PHASE. Int J Med Sci. 7(2):72-81.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.